Introduction
Endometrial receptivity and implantation failure remains an unsolved enigma of the 20 million years of human life on our planet. Way of life, work or occupation, eating habits, relationship with partner, exposure to environmental toxins, physical activities etc., influence endocrine, psychological, immunological systems, changing the genome and influencing endometrial receptivity. Endometrial receptivity is orchestrated by the central nervous system cortex, all endocrine glands and is regulated by estrogens, progesterone, other hormones, as well as autocrine and paracrine factors. Endometrial receptivity is the ability of the uterus to accept and develop a new embryo [1]. Guzeloglu-Kayishi O, et al. [2] reported many hormones (steroid and non-steroid) receptors, growth factors and cytokines required for implantation.
Successful implantation requires coordination of embryo development and receptivity of the endometrium. The mother-embryo signaling pathway is highly complex. High molecular weight fractions, including proteoglycans, mucins and albumin, are produced in association with implantation and sequestered within are a host of lower molecular weight (<1000 KDa) proteins, including endocrine hormones, immunoglobulins, growth hormone-related peptides, immunomodulatory peptides (cytokines, chemokines, interferones, proteases) [3]. They are secreted by luminal and glandular epithelium under local and systemic endocrine control regulated by the hypothalamic-pituitary-gonadal axis. Integrins are the best characterized immunohistochemical markers of uterine receptivity. One of the first embryo-specific protein, reported to be secreted, at the blastocyst stage, by the human embryo, is human chorionic gonadotropin, which is involved in mediating the immune response. The yolk sac, one of the earliest organs in the human embryo, starts in the preimplantation blastocyst as part of the extra-embryonicmesoderm and contributes to both the developing embryo and the trophoblast. It acts as a bridge between the free-living stages of the embryo’s life and the establishment of placental nutrition and respiration. Without these components there is fetal wastage, congenital abnormalities and likely long-term postnatal diseases.
After the blastocyst (0.2 mm diameter) attaches to the endometrium in a region of increased pinopode expression, a complex cascade of cytokines and chemokines, morphogens, adhesion molecules, hormones, and transcriptional and growth factors takes place to facilitate implantation [4].
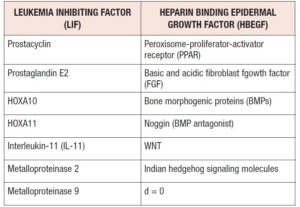
Macrophages produce cytokines (LIF, IL-11) essential for the implantation. Cytokines are regulated by ovarian steroids. They mediate embryo growth, differentiation and immune-related implantation. IL-11 mediates trophoblast invasion and its deficiency is related to a reduction in endometrial natural killer (NK) cells. In the window of implantation (WOI), NK cells are the most represented immune cells,being critical regulators of angiogenesis, immunotolerance and trophoblast invasion [5].
Four major phases of endometrial transformation make transcriptomic signature in luminal-glandular epithelium leading to abrupt transcriptomic opening of the WOI at single cell level. Decidualization is initiated before the opening of the WOI and direct interplay between stromal fibroblasts and lymphocytes is found during decidualization [6]. Human chorionic gonadotropin (hCG), produced by the blastocysts, prolongs the WOI by inhibiting endometrial insulin-like growth factor-binding protein -1 (IGFBP-1) production. It augments angiogenesis at the implantation site by increasing vascular endothelial growth factor (VEGF) expression, modulating local cytokine and chemokine expression, local protease activity and invasion potential. Preimplantation factors (PIFs) were secreted in 77.6% of all successful implantations. All PIF-negative embryos failed to implant (Table 1).
Estradiol and progesterone
Estradiol and progesterone are the basic hormones regulating endometrial receptivity. Cyclical ovarian-derived steroid hormones are central for programming cellular responses and normal endometrial function. The master regulators of pregnancy success are the endometrial stromal fibroblasts (eSFs). Estradiol “extensively affected eSF DNA (deoxyribonuclear acid) methylome and transcriptome. [Estradiol] resulted in a more open versus closed chromatin, confirmed by histone modification analysis” [7]. Steroid hormones regulate genes in the endometrium. The estradiol/progesterone ratio plays a critical role in this. If the hormone response is abnormal, infertility can result. Interaction of steroid hormones with a second genome (epigenome) to regulate gene expression is a mechanism not fully understood. Important genes influencing successful implantation include: connexin 43 (Cx43), dickkopf 1, glycodelin, homeobox A10, Integrin α V β 3, interleukin-1 β (IL-1β), leukemia inhibiting factor, progesterone receptor β, prolactin, retinol binding protein 4, transducer of Erb B2, and vascular endothelial VEGF-A [8]. The “decidual“ program includes upregulation of stromal ER-α, PRs, Cx43 gap junction proteins. Also, ovarian hormones regulate ion channels involved in the regulation of uterine electrolyte and fluid transport, promoting decidualization and regulation of genes associated with the process of implantation. Ca 2+ channels may play a role in blastocyst adhesion in the endometrium [9].
Progesterone modulates maternal immune responses (protection of the semiallogenic fetus), improves utero-placental circulation and vasodilatation, decreases apoptosis, promotes extravillous trophoblast invasion in the maternal decidua, remodeling the local vasculature, suppresses the fetal immunoplacental inflammatory response, decreases uterine contractions, and induces cervix ripening [10]. Keratinocyte growth factor (KGF), derived from uterine stromal cells, is upregulatd by the action of progesterone. When progesterone and KGF2 levels drop, the blood supply of the endometrium and muscle layers decreases [11]. Progesterone protects ovarian function against ischemic reperfusion injury through antiapoptotic and antioxidative properties [12].
Glucose/insulin
Insulin is detected at the 4-cell blastocyst stage. Even ordinary but ill-timed excessive glucose intake could be a cause of implantation failure, spontaneous pregnancy loss and congenital abnormalities [13,14]. Primary signaling pathways include the insulin/insulin-like growth factor (IGF), TOR, and sirtuin networks which alter mitochondrial function and metabolic activity via genome proteostasis [15]. Hyperinsulinism decreases the number of insulin receptors and IGF receptors, as well as levels of IGFBP-1 and sex hormone-binding globulin (SHBG). The hyperinsulinism that is a consequence of insulin resistance (IR) involving plasminogen activator inhibitor-1 (PAI-1), which inhibits plasminogen activator and subsequent fibrinolysis, has potential thromboembolic effects. Increased androstenedione (induced by hyperinsulinism) inhibits cell growth and activity. Glycodelin secretion inhibits endometrial immune response to the embryo. Increased testosterone decreases HOXA10 (essential for the implantation). IGFBP-1 facilitates adhesive progress at the fetoplacental interface. Also, insulin is able to act centrally, modulating the centers involved in reproduction: the KYDN neurons and gonadotropin releasing hormone (GnRH)-secreting neurons [16]. Dodd et al. [17] showed that glucose, insulin, leptin, ghrelin, adiponectin and androgens influenced GABA, which regulates apetite and estradiol balance. Hyperinsulinism can be one of the causative factors for anovuation, and lower progesterone levels in the luteal phase and in the first trimester of pregnancy.
Genazzani et al. [18] showed that administration of acetyl-L-carnitine, L-carnitine, L-arginine and N-acetylcysteine improved hyperinsulinism, supporting the hypothesis that liver function is impaired in hyperinsulinemic women with polycystic ovary syndrome (PCOS). Morin-Pepunen et al. [19] described improved expression/synthesis of the hepatic insulin degrading enzymes (IDEs), which are responsible for at least 70-80% of insulin clearance. Carvalho et al. [20] confirmed that L arginine improves insulin sensitivity in beta cell function in the offspring of dibetic rats through Akt and PDX-1 activation. Both L-arginine and N acetyl carnitine donate thiol groups, while reactive oxidative glutathione improves nitric oxide synthesis leading to protection of endothelial cells and improving insulin sensitivity [21].
The carnitines, as well as other components, reduce the effects of the free radicals, and can stabilize the genome as well as the cellular membranes. A study by Varnagy et al. [22] showed that a state of carnitine deficiency may be related to the numbers of ovum produced during IVF procedures. The study examines the levels of the carnitines in the follicular fluid and whether the state of deficiency can be prevented. Virmani et al. [23] also looked at strategies to reduce the issues of aneuploidy. They showed that intra-follicular ischemia and hypoxia negatively influenced spindle organization and chromosomal segregation in the human oocyte and that this can be reduced by the L-carninitine, L- arginine, inositol formulation.
Energy metabolism in cells is altered when hyperinsulinism is treated with metformin, as this lowers glucose levels by inhibiting hepatic neoglucogenesis and opposing actions of glucagon, and by inhibiting IGF-1 insulin signaling through AMPK-dependent phosphorylation of IRS-1, which transmits signals from insulin and IGF-1 receptors to the PI3K-AKT pathway. The inhibition of mitochondrial complex 1 of the electron transport chain induces a drop in energy charge, resulting in adenosine triphosphate decrease (ATP), adenosine monophosphate (AMP) increases binding P-site adenylate cyclase enzyme and inhibition of activity leading to defective cAMP protein kinase A (CAMPK) signaling on glucagon receptor [24]. AMPK, an energy sensor, is a master coordinator of an integrated signaling network that comprises metabolic and growth pathways and works to restore cellular energy balance, switching on catabolic pathways that generate ATP and switching off anabolic ATP-consuming pathways. Metformin can induce weight loss, reducing PAI-1; it also inhibits platelet aggregation, reduces inflammatory cytokine level and adhesion molecules ICAM1 and VCAM [25], decreases androgens, IR, glycodelin, IGF-1 protein expression and improves endometrial function and receptivity.
Bearing in mind the detrimental effects of hyperinsulinism on endometrial receptivity, we suggested that the oral glucose tolerance test (OGTT) be routinely performed at 8 a.m. by oral ingestion of 75 gr of glucose, with glycemia and insulin detection performed 0, 30, 60, 90 and 120 minutes later. The area under the curve has to be calculated because the HOMA (homeostatis assessment) index has a very limited value. Hyperinsulinism and IR have to be treated by an adequate metformin dose at least 3-6 months preconceptionally or later, during the pregnancy, with a proper dietary regimen, in order to avoid miscarrigies [26].
Cortisol, prolactin and stressors
Stressors increase levels of corticoreleasing hormone (CRH), adrenocorticotropic hormone, cortisol and prolactin. Increased CRH supresses GnRH pulse secretion, decreasing follicle-stimulating hormone (FSH), luteinizing hormone (LH) and estradiol. Hypoestrogenism affects contraction of the uterine basal and spiral arteries, followed by a rise in a peripheral vascular resistance. Stressors decrease PGE2 release, and vasoconstriction potentiated by angiotensin II, nitric oxide, acetylcholine and serotonin [27]. The systemic sympathetic adrenomedullary system is involved (increased norepinephrine, CRH, IL-6), dyscoagulation and inflammation. The gene ontologies of stress-repressed genes strongly suggested that stress influences expression of genes related to the metabolome, which include the leptin receptor gene. Similar to LIF, leptin is a pleiotropic and ubiquitous cytokine that plays a critical role in the reproductive function and increases levels of LIF and its receptors. It is produced and secreted by blastocysts. Calmodulin, the primary mediator of Ca 2+-dependent signaling, enhances the stability of the estrogen receptor (ER). An association was found between repressed response to estradiol and reduction of leptin signals and calmodulin. Stressors altered uterine gene expression through an ovarian-independent pathway resulting in decreased uterine receptivity [28]. The mTOR serine/threonine protein kinase pathway regulates cell growth and proliferation. Rapamycin, an inhibitor of TOR and a macrolide immune suppressor, inhibits activation of follicles by regulation of the mTOR/sirtun signaling pathway, protects ovarian reserve, and extends the reproductive age [29].
Zhao et al. showed that retraint-induced stress inhibits mouse implantation by impairing uterine receptivity and downregulation of estradiol, progesterone and heparin-binding epidermal growth factor [30]. Dong et al. found that fertility stressors (depression, anxiety, sleep disturbances) negatively affected sex hormone and neuroimmmunological function [31]. The findings of this study were confirmed by those of Engert et al., who showed that stressors can excite catecholemine alpha receptors causing vasoconstriction and reducing blood flow [32]. In a group of 300 infertile women, they found reduced endometrial and subendometrial flow due to changes in the hypothalamus-pituitary-adrenal axis and the sympathetic adrenomedullary system. Increased levels of glucocorticoids and catecholamines were confirmed by Taskewi et al. [33]. Fertility stressors are not associated with endometrial thickness. Endometrial receptivity is decreased due to lowered endometrial flow. Cortisol concentration can be elevated in the endometrium, whereas 11 β hydroxysteroid dehydrogenase (HSD) 2 expression is diminished. In endometrial biopsies performed in PCOS patients with IR, Qi et al. found that cortisol-attenuated insulin-stimulated glucose uptake in EECs was mediated by inhibition of Akt phosphorylation and glucose transporter type 4 translocation via induction of phosphatase and tensin homolog deleted on chromosome ten (PTEN) [34]. Decreased oxidation of cortisol and defects of insulin signaling in the endometrium were observed in PCOS with IR. The exessive cortisol level derived from the reduction of 11 β HSD2 might contribute to the development of endometrial IR by inhibiting the insulin signaling pathway via induction of PTEN expression. Their in vitro study suggested a detrimental role of cortisol on insulin sensitivity in the endometrium. 11 β HSD2 only has an oxidase function, converting active cortisol to inactive cortisone [35]. Some studies have linked endometrial IR to decreased endometrial receptivity and tumorigenesis [36]. In infertile women, Wdowiak et al. showed higher levels of cortisol and prolactin, decreasing preovulatory LH peak and postovulatory estradiol [37].
Growth hormone
The major growth factor family includes the epidermal growth factor (EGF), fibroblast growth factor, insulin growth factor (IGF), tissue growth factor, and heparin-binding growth factor, as well as amphiregulin and neuregulin, members of the EGF family.
Growth hormone (GH) has been shown to improve in vitro fertilization due to its stimulatory effects on oocyte quality. Additional positive effects can be seen on endometrial receptivity [38]. A receptive endometrium is favorable for embryo adhesion and the subsequent attachment and invasion process. GH and insulin-like growth factor I (IGF-1) are expressed in the endometrium. Lower stromal expression of GH in «luteal phase defect» (progesterone below 8 ng/ml) delayed endometrial maturation. GH, both directly and in an IGF-I mediated manner, induces human endometrial cells to promote proliferation, vascularization and up-regulation of receptivity-related genes such as VEGF and integrin β 3 (biomarkers of receptivity) [39]. VEGF is important in angiogenesis, acting in an autocrine manner on endometrial epithelial cell adhesion as a key regulator in the implantation process. GH has stimulatory effects on ovarian steroidogenic cell function and maintaince of the corpus luteum. Dakhly et al. showed that addition of 7.5 IU/day of GH from day 6 of HMG until the day of hCG triggering improved fertility rate in women over 45 years of age and with FSH>20 IU/L [40], whilst Liu’s study in women 20-40 years of age and with poor quality embryos treated them with 2-4 IU/daily from day 2 of the previous cycle (6 weeks GH pretreatment) [41]. Liu et al. showed that rGH treatment of mice with premature ovarian insufficiency reduced premature ovarian insufficiency (POI) histopathology in ovarian tissue, relieved ovarian granulosa cell injury, reduced the number of atretic follicles and increased the number of mature oocytes [42]. GH may promote ovarian tissue repair, estradiol release and oocyte maturation via activation of the Notch-1 signaling pathway in ovarian tissue. The Notch signaling pathway has a critical role in the development and homeostasis of tissue by regulating pathology, proliferation, differentiation, apoptosis and stem cell self-renewal.
Melatonin
Circardian rhythm dysregulation followed by low melatonin is associated with a low implantation rate [43]. Melatonin receptors 1A and 1B are localized by immunohistochemistry in glandular epithelial cells on endometrial biopsies [44]. Melatonin promotes mitochondrial homeostasis by regulating molecular DNA and mtDNA transcriptional activities. The suprachiasmatic nucleus in the hypothalamus coordinates the clock mechanism in peripheral tissue (lung, heart, kidney, pancreas, non-pregnant uterus). The cellular clock oscillates depending on daily timing through autoregulation transcriptional/translational feedback loops in which the heterodimer BMAL1/ CLOCK drives the expression of period (Per) and cryptochrome genes. Loss of Per 2 expression by siRNA knock down perturbed circardian oscillations in decidualizing human endometrial stromal cells, affecting mitotic expansion by blocking the G2/M phase, which is positively associated with misscarriages. Melatonin treatment attenuated estradiol-induced endometrial epithelial cell proliferation in culture. Also, melatonin therapy decreases pain score. With its autocoid, chronobiotic, hypnotic, immunomodultive factors and biological modulator capacity melatonin improves fertility rate in women with premature ovarian insufficiency [45].
DHEA
DHEA affects the activity of 21 hydroxylase and 11 β hydroxylase, responsible for cortisol synthesis from precursors 17 OH progesterone and 11-deoxycortisol. Dehydroepiandrosterone (DHEA) is a peripheral estrogen and androgen precursor, adrenal enzyme regulator, 5 α reductase stimulator, pituitary beta endorphin stimulator, pituitary sensitizer and neurosteroid [46]. It increases lipolysis and glucose uptake, and decreases adipocyte differentiation, adipocyte tissue mass and 11 βHSD1 activation. Increased cortisol/DHEAS has been found to potentiate metabolic syndrome, obesity, diabetes mellitus, osteoporosis, neurodegenerative disorders and cardiovascular diseases. DHEA increases T lymphocyte infiltration, resulting in a decline of CD4+TLy and upregulation of CD8+TLy. DHEA can enhance the T helper 1 immune response and regulates balance of the Th1/Th2 response. The products of Th1 are interferon γ and TNF. It regulates antigen presentation and immunity against intracellular pathology. DHEA treatment can increase selective T Ly infiltration in mice, resulting in decline of the CD4+T Ly population and upregulation of the CD8TLy population [47]. DHEA is converted into estradiol which suppresses FSH. DHEA increases testosterone production by the very early follicles stimulating androgen receptors, allowing more preantral follicles to progress to more mature antral follicles [48].
Our study performed in 820 women with POI showed lower DHEAS in a group aged 30-40 years, compared with women 20-30 years old. This represents an additional factor influencing lower endometrial receptivity rate [49].
Thyroid gland hormones
Thyroid hormones increase lipid metabolism, thermogenesis, and lipolytic activity due to increased beta 2 adrenergic receptor expression and cAMP-activated hormone-sensitive lipase activity. Thyroid-releasing hormone directly affects the ovaries, increases prolactin and thyroid-stimulating hormone (TSH). TSH receptors are present on immature oocytes [50]. Higher postovulatory estradiol levels decrease thyroxin and increase TSH, in stimulated cycles (ovulation induction). In hypothyroidism, GnRH secretion and peripheral estradiol metabolism are disturbed, pulsatility of LH is abnormal, prolactin is increased, and hemostasis is defective. Free steroid fractions, excretion of 2-oxyestrogens and peripheral aromatization are increased [51].
In order to improve endometrial receptivity, TSH levels of 1-2.5 mmol/L are advised for at least 3-6 months preconceptually.
Adenomyosis
Endometrial receptivity and implantation failure in adenomyosis can be induced by: aberrant endometrial metabolism (altered endometrial steroid metabolism, abnormal inflammatory response, alteration of ER and PR expression), altered uterine oxidative stress environment (abnormal levels of free radicals, low oxygen), lack of expression of adhesion molecules (integrin, cadherin, selectin), reduced expression of implantation markers (LIF, NF-kBm IL-6) and altered function of the genes for embryo development (HOXA10). The HOXA genes influencing the development of Mullerian ducts are: HOXA9 oviduct, HOXA10, HOXA11 uterus, HOXA13 cervix, and HOXA13 upper vagina. Exposure of the embryo to thalidomide, infectome agents or ionizing radiation affects the uterine morphology by triggering changes in both the location and amount of HOXA expression [52].
Endometriosis
“The internal pelvic organs receive nerve supply from the autonomic nervous system, sympathetic, and parasympathetic nervous system. Autonomic T11 and T12 innervate the uterus, and it derives its sympathetic nerve supply from the hypogastric plexus, and the parasympathetic supply is from S2 to S4” [53].
Endometriosis is an inherited, autoimmune life-long disease [26]. In order to achieve pregnancy, endometriosis has to be treated continuously from the time of diagnosis confirmation until the time of ovulation induction.
As reported by others “[progesterone] responses were abberant in early and late stage endometriosis, and mapping differentially methylated CpG sites with progesterone receptor targets from the literature revealed different but not decreased [progesterone targets], leading to question the [progesterone]-“resistant” phenotype in endometriosis. Interestingly, aberrant [estradiol] response was noted in eSF from endometriosis women; (...) Steroid hormones affected specific genomic contexts and locations, significantly enriching enhancers and intergenic regions and minimally involving proximal promoters and CpG islands, regardless of hormone type and eSF disease state; (...) In eSF from women with endometriosis, aberrant hormone-induced methylation signatures were mainly due to existing DNA methylation marks prior to hormone treatments and involved known endometriosis genes and pathways. (...) Distinct DNA methylation and transcriptomic signatures revealed [that] early and late stage endometriosis comprise unique disease subtypes” [7]. Hormone-epigenome-transcriptome interplay of each steroid hormones was detected in normal SF and aberrant estradiol response. Moreover, environmental and inflammatory signals can alter steroid hormone-driven endometrial epigenome.
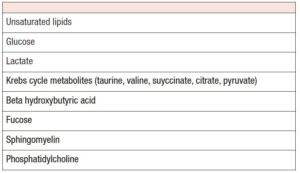
Chronic inflammation affects the chromatin landscape in endometriosis [54]. Morelli et al. found lower levels of acetate citrate, beta hydroxybutyrate and valine in women with endometriosis [55].
Inflammatory mediators impair decidual function. Estradiol, progesterone and cAMP, mediated by connexins, influence prolactin and IGFBP-1. IL-1β produces classical endometriosis symptoms, pronounces angiogenesis (VEGF) and invasiveness, and reduces Cx43 expression via ERK1/2 pathway [56]. ERK inhibition stimullates decidualized morphology (Table 2).
Machairiotis et al. found increased monocyte chemotaxis, neurogenesis and fibrosis [57]. NF-kB is prominent in the IL-1β signaling pathway. Pluchino et al. found a lack of HSD17β2 expression in endometriosis [58]. Progesterone does not blockade aromatase expression.
Xu et al. showed that oocytes from women with mild endometriosis exhibited abnormal mitochondrial structures and decreased mitochondrial mass [59]. Glutathione peroxidase 3 and thyoredoxin binding protein 2 negatively correlated with the percentage of mature oocytes. Other authors reported a possible imbalance in the thiol-redox system and increased levels of inflammatory cytokines in the intrafollicular microenvironment that may affect the embryo [60].
Oxidative stress is induced by the imbalance in the rate of function and removal of free radicals in patients with endometriosis. Giorgi et al. found positive effects of antioxidants,
L-carnitine and N-acetylcysteine in preventing meiotic oocyte damage [61]. Excessive production of the autoantibodies to the endometrium impairs implantation rate and endometrial receptivity. Increased levels of the following factors have been detected: IL-6, IL-8, TNFalpha, atilaminin 1 antibodies, zona pellucida antibodies, beta-2-glycoprotein, cardiolipin, ethanolamine, thyreoredoxin-binding protein 2, arrested embryo, superoxide dismutase 1 in cumulus cells [62].
Autoimmune causes
Endometrial receptivity failure and abortions are thought to be caused by the presence of autoantibodies, already existing in women, against cell membrane, phospholipids, thyroid antigens, nuclear Ag, syncytiotrophoblast cells or against other organelles or tissues. About 10% of the total are antinuclear antibodies, regardless of medical history of reccurent pregnancy loss.
Antiphospholipid antibodies are a family of immunoglobulins that react with anions of phospholipids or anions of phospholipid-protein complexes in the cell membrane of the syncytiotrophoblast. They induce vessel thrombosis of the surrounding placental maternal unit, placental infarction and fetal death. The primary mechanism in the first trimester depends on deleterious effects directly on trophoblastic cells, inhibition of secretion of human placental chorionic gonadotrophin, and expression of trophoblast cell adhesion molecules (a1, a5 integrins, E, VE-cadherins).
Possible actions of antiphospholipid antibodies are: abnormalities in endothelial cell function vessels, obstructive angiopathy, platelet stimulation and/or adhesion, placental infarction, inibition of protein C and stimulation of protein S. They inactivate clotting factors Va and VIIa. Their stimulation increases coagulation, and reduces annexin V, a protein with potent antithrombotic effects on the surface between the trophoblast and the endothelial cells. Phospholipids bind to the surface of trophoblasts and cause direct destruction of cells, inhibition of syncytiotrophoblast formation, decrease of Hcg and defective penetration into maternal tissue.
Increased levels of CD 56+ Ly and NK cells are detected in the secretory phase. Reccurent abortions are associated with immunity type Th1 (IF-γ)+, TNF and IL-12.
Pregnancy and coagulation
Increased procoagulant factors and decreased anticoagulant factors (naturally occuring anticoagulation, reduction of fibrinogenolysis, modified maternal response to hemostasis) may lead to malfunction in the fetoplacental unit. Familial thrombophillia (factor V Leyden mutation, acquired activated protein C resistance, antiphospholipid antibodies syndrome and hypofibrinolysis) contributed to implantation failure and early pregnancy loss [63]. The infiltration of trophoblasts in arcuate arteries is essential for implantation, placentation and regular continuation of pregnancy. Thrombosis of spiral arteries and the intervillous space on the maternal side of the placenta can impair placental perfusion [64].
5,10-methylenetetrahydrofolate reductase catalyzes the conversion of 5,10 methylentetrahydrofolic acid to 5-methyltetrahydrofolic acid (this takes part in the methylation of homocysteine in methyonine). Substitution of the cytosome molecule by a thymine molecule at position 677 increases the incidence of homocysteine and thrombophilia. Reduced MTHFR activity and hyperhomocystinemia are clinically manifested when lack of folic acid coexists.
Elevated PAI activity is detected in genetic and metabolic disorders, IR, hypertension and smoking. PAI-1 is the major physiological inhibitor of plasminogen activator, and it plays a central role in fibrinolysis. Polymorphism of the PAI-1 gene is associated with high levels of PAI and reduced fibrinolytic activity. 4G homozygosity has been associated with the complications such as implantation failure, preeclampsia, prenatal intrauterine growth, placental death.
Diagnostic procedures in clinical practice
- Day 2-5 of menstrual cycle: FSH, LH, estradiol, AMH, inhibin B, prolactin, cortisol, GH, DHEAS, testosterone, androstendione, 17 OH progesterone, fT4, TSH, vitamin D, karyotype, anticardiolipin Ab, beta 2 glycoprotein Ab, thyroglobulin Ab, thyroperoxidase Ab, lupus anticoagulant, homocysteine, PAI, MTHFR, factor V Leyden, prothrombin G20210A.
- OGTT with 75 gr of glucose ingestion at 8 a.m. after 12-hour starvation. After glucose ingestion, blood samples are taken immediately and then at half hourly intervals during 2 hours, to measure glucose and insulin.
- 21 and 24. Day of cycle: estradiol and progesterone.
Advised therapy approach
- Full understanding, psychological support and empathy;
- Natural progesterone in a case of insufficient luteal phase (200 mg orally or vaginally at bedtime from day 14-25th day of the cycle)
- Estroprogestogens for women with ovarian insufficiency (premature ovarian failure, early menopause, etc.)
- Dopamine agonists for hyperprolactinemia
- Prednisolone 10-20 mg/day: may prevent recycling in the circulation of cardiolipin or suspend the discharge of embryotoxic factors or factors associated with HLA; lowers NK (CD 56+/CD16+) cells.
- Aspirin 100 mg should be given preconceptionally. It may suspend cyclooxygenase action on platelets by suspending the composition of thromboxane thrombosis and thus preventing vascular thrombosis in placental blood vessels.
- Heparin: heparin of low molecular weight prevents chorionic villous sampling phospholipids from being destroyed by assisting in the successful implantation in the early stages of pregnancy.
- Ca: 500 mg, vitamin D 1000 mg
- Metformin: decreases androgens, improves endometrial function and implantation, decreases IR; glycodelin and IGF-1 protein expression is corrected.
- Folic acid: 0.5-2 mg for normalizing homocysteine level
- L-arginine, L-carnitine, acetyl-L-carnitine, N-acetylcysteine
- Melatonin 2 mg at bedtime
- Anxiolytics in low doses
- Sildenafil, a selective inhibitor of 5-phosphodiesterase, an enzyme included in cGMP hydrolization, decreases NK cell activity [65]. Vaginal suppositories 25 mg every 6 hours is suggested in the proliferative phase [66].
- Intralipid 20% intravenously (9 mg/ml total blood volume) corresponds to 2 ml of intralipid 20% diluted in 250 mg saline [67].
Conclusion
The complex interplay between genetic, environmental, endocrine, immunological, psychological and hematological parameters needs to be clarified and tested in order to arrive at a more complete understanding of the etiology of impaired endometrial receptivity.
A complete diagnostic procedure is needed. It is advisable to treat all detected irregularities for at least 6 months preconceptionally. Prevention of many diseases later in life starts with detection of the etiological factors underlying impaired endometrial receptivity and therapeutic improvement of the same.
Disclosure statement
No potential conflict of interest was reported by authors.