Introduction
Perimenopause is a midlife endocrine aging transition in women characterized by menstrual cycle irregularity caused by fluctuations of hypothalamic, pituitary, and ovarian hormone levels.1,2 During perimenopause, 80% of women experience multiple climacteric symptoms such as insomnia, hot flushes, depression and memory impairment.3 There is a general consensus that the changes in energy metabolism in the brain that accompany the endocrine transition result in a state that is prodromal of several pathologies, including cardiovascular disease (CVD)4 and dementia-related disorders, especially Alzheimer’s disease (AD) which has a high prevalence during the subsequent postmenopausal phase, particularly at 15-20 years after menopause. 1,5-7 Indeed, several studies report menopause-associated alterations in systemic lipid and glucose metabolism, resulting in increased central fat distribution, increased blood glucose and insulin levels, as well as the appearance of an atherogenic lipid profile.8,9 Moreover, the decline in estrogen levels has been associated with a rise in total cholesterol, low density lipoprotein (LDL) and triglyceride levels with a concomitant reduction of high density lipoprotein (HDL),8,9 indicating that altered hormone levels peculiar to the perimenopause expose women to an increased risk of CVD. In addition, the disruption of several estrogen-regulated systems in the brain can increase the risk of developing age-related neurological disorders.10,11 Epidemiological studies show that almost two-thirds of AD patients are women and that hormone replacement therapy (HRT), when started before menopause, may protect females from dementia, suggesting that endocrine-induced physio-metabolic changes during perimenopause influence the cognitive trajectory in female brains.12-14 Therefore, the metabolic changes of perimenopause occur not only in the periphery, but also in the brain, resulting in particular vulnerability for women in this phase of life.
Recent studies demonstrate that metabolic changes in the brain occur during the perimenopausal transition, with a decline in glucose uptake leading to ketone body dependence and later, to the utilization of lipids of white matter origin as a secondary energy source.5,14,15 These changes also occur in the Alzheimer’s brain.11,15 Alterations in brain cholesterol metabolism have been associated with normal aging, as supported by consistent reports of impaired synthesis and transport of lipoprotein involving apolipoprotein E (ApoE) and cholesterol 24-hydroxylase (CYP46).16 Considering that cholesterol is a major structural and functional component of myelin sheaths, synaptosomal membranes, lipid rafts, intracellular vesicles and synaptic connections,17-19 alterations in brain cholesterol metabolism and transport could affect synaptic plasticity and cognition, known to be reduced in AD patients.16,20
In the present study, we evaluated changes in genes and proteins involved in brain cholesterol homeostasis in a rat model of human perimenopause. In addition, considering that cholesterol metabolism in the CNS is highly specialized and compartmentalized, with tightly regulated cross-talk with the periphery, we also investigated possible association between brain cholesterol metabolism and serum lipid metabolites in our rat model and in a cohort of women.
Methods
Animal model
Animal studies were performed according to the National Institutes of Health guidelines on use of laboratory animals; protocols were approved by the University of Southern California Institutional Animal Care and Use Committee. As previously reported,2 Sprague-Dawley female rats aged 9-10 months old (mo) were assigned to groups according to their stage of ovarian senescence, established on the basis of the Stages of Reproductive Aging Workshop (STRAW) bleeding criteria that characterizes human reproductive aging through menopause.2,21 The endocrine aging groups included: regular cyclers (4-5 day cycles), irregular cyclers (5-8 day cycles), and acyclic (no cycling within 9 days) at 9-10 mo. Six mo regular cycling rats and 16 mo acyclic rats were used as controls for chronological aging before and after the perimenopausal transition, respectively.22 A total of 23 animals were utilized.
Rat brain tissue collection
Rats were euthanized and the brains were rapidly dissected on ice. The cortical left hemisphere was fully peeled laterally, harvested, and frozen at -80 °C for subsequent analyses.
Lipoprotein signaling and cholesterol metabolism gene expression array
Lipoprotein signaling and cholesterol metabolism RT2 Profiler PCR array (Qiagen, Hilden, Germany) was performed on 84 key genes involved in lipoprotein transport and cholesterol metabolism. Total RNA was isolated from rat cortex tissue using the PureLink Mini Kit (Invitrogen, Waltham, MA USA). The quality and the quantity of RNA were analysed before complementary DNA (cDNA) synthesis using RT2 First Strand kit (Qiagen, Hilden, Germany). SYBER®-Green real time and quantitative polymerase chain reactions (RT-PCR), were performed on 384-well plate, with 10 ng cDNA samples mixed with RT2 SYBER®-Green master mix and the fluorescence was detected on an ABI 7900HT Fast Real-Time PCR System (Applied Biosystems, Waltham, MA USA). Data were analysed using RT2 Profiler PCR Array Data Analysis version 3.5 (SABioscence-Qiagen, Hilden, Germany). Relative gene expression levels or fold changes relative to the reference group were calculated by the comparative Ct (ΔΔCt) method, with Ct denoting threshold cycle. For each sample, ΔCt was calculated as the difference in average Ct of the target gene and the endogenous control gene. For each group of 4 samples, mean 2-ΔCt was calculated as the geometric mean of 2-ΔCt of all samples in the group. Fold change was then calculated as mean 2-ΔCt (comparison group)/mean 2-ΔCt (reference group). The 2-ΔΔCt values for each target gene were statistically analysed by ANOVA followed by pairwise comparisons using Student’s t-test. The statistical significance was determined as p<0.05; p-values were not adjusted for multiple testing as discussed previously.23
Bioinformatic analysis by Ingenuity Pathway Analysis
Expression data for genes were analysed by Ingenuity Pathway Analysis core (Qiagen, Hilden, Germany) composed of a network analysis (cut-off settings: fold change 1.2; p-value 0.05). The network analysis identified biological connectivity among molecules in the data set that were relatively up-regulated or down-regulated and their interactions with other molecules present in the Ingenuity Knowledge Base (IKB). Focus molecules were combined into networks that maximized their specific connectivity (direct and indirect interactions among focus and interacting molecules). Generated networks were ranked by the network score according to their degree of relevance to the network eligible molecules from the data set. The network score was calculated with Fisher’s exact test, taking into account the number of network eligible molecules in the network and the size of the network, as well as the total number of network eligible molecules analysed and the total number of molecules in the IKB that were included in the network. Higher network scores are associated with lower probability of finding the observed number of network eligible molecules in a given network by chance. Ingenuity’s Upstream Regulator Analysis in Ingenuity Pathway Analysis is a tool that predicts upstream regulators from gene expression data based on the literature and compiled in the IKB. A Fisher’s exact test p-value was calculated to assess the significance of enrichment of the gene expression data for the genes downstream of an upstream regulator. A z-score was given to indicate the degree of consistent agreement or disagreement of the actual versus the expected direction of change among the downstream gene targets. A prediction about the state of the upstream regulator, either activated or inhibited, was made based on the z-score.
Protein expression analysis
Brain cortex tissue was stored at -80°C and was processed for protein extraction using Tissue Protein Extraction Reagent (Thermo Scientific, Rockford, IL USA) with phosphatase and protease inhibitors (Sigma, St. Louis, MO USA). Protein concentrations were determined with Bradford assay (Bio-Rad Laboratories, Hercules, CA USA). Equal amounts of protein (20 µg/well) were loaded to SDS PAGE Criterion gel (Bio-Rad, Hercules, CA USA) and electrophoresed with Tris/glycine running buffer (pH 8.3). Proteins were transferred to 0.45-µm pore size polyvinylidene difluoride membranes and probed with primary antibodies against: Liver x Receptor β (LXR beta/NR1H2, Novus Biologicals USA, Littleton, CO USA), cholesterol 24-hydroxylase (cyp46, Millipore, Darmstadt, Germany), Lipoprotein repector-11 (LR11, BD transduction laboratories, Franklin Lakes, NJ USA), scavenger receptor class B member 1 and ATP binding cassette 1 (ABCA1), apolipoprotein E (ApoE), 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), low density lipoprotein receptor-related protein 1 (LRP1), low density lipoprotein receptor (LDLR), 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2). Primary antibodies against ABCA1, ApoE, HMGCR, LRP1, LDLR, and HMGSC2 were obtained from Abcam (Cambridge, MA USA). Horseradish peroxidase-conjugated anti-rabbit antibody, horseradish peroxidase-conjugated anti-mouse antibody (Vector Laboratories, Burlingame, CA USA) and horseradish peroxidase-conjugated anti-goat (Abcam, Cambridge, MA USA) were used as secondary antibodies. Immunoreactive bands were visualized with Pierce SuperSignal Chemiluminescent Substrates (Thermo Scientific, Waltham, MA USA) and captured by Molecular Imager ChemiDoc XRS System (Bio-Rad Laboratories, Hercules, CA USA). Band quantification was performed using Image Lab software (Bio-Rad Laboratories, Hercules, CA USA). Protein levels were normalized to expression of β-actin as a loading control.
Human cohort
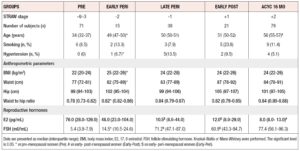
The study population consisted of a randomly selected sample of women attending the Menopause and Osteoporosis Centre of the University of Ferrara (Ferrara, Italy). This study was designed and performed according to The Code of Ethics of the World Medical Association (Declaration of Helsinki) and was conducted according to the guidelines for Good Clinical Practice (European Medicines Agency). Written informed consent was obtained from each patient before inclusion in the study. Study participants (n=224 Caucasian women aged 21-67 years) were in endocrine states characterized according to STRAW +10 criteria23 at the time of enrollment. The presence of amenorrhea and the levels of serum follicle stimulating hormone (FSH) were used to assign a STRAW status to each woman. Serum levels of FSH and E2 estradiol were assessed using commercially available chemiluminescent microparticle immunoassays (Architect, Abbott Park, IL USA). Women under hormonal treatment (including vaginal estrogens) and women diagnosed with chronic disease (e.g. diabetes, hypertension, etc.) were excluded from the study.
Cholesterol, triglycerides and ketone body assays in serum
Concentration of total cholesterol, triglycerides, and cholesterol-HDL were determined enzymatically using commercially available kits (Randox Laboratories, Crumlin, UK).
Total cholesterol (Tot-Chol) was determined enzymatically after hydrolysis of cholesteryl esters by cholesterol esterase and oxidation of the resulting free cholesterol by cholesterol oxidase. The quinoneimine produced by the reaction of resulting hydrogen peroxide and phenol was detected spectrophotometrically at 505 nm. Final concentrations were calculated by interpolation with a standard curve of cholesterol provided with the kit (Randox Laboratories, Crumlin, UK; cat. CH200).
HDL-cholesterol (HDL-C) was assessed on serum samples pre-treated with phosphotungstic acid (0.55 mM) that, in the presence of magnesium ions (25 mM magnesium chloride), induce the precipitation of LDL, VLDL and chylomicrons fraction. After centrifugation for 10 minutes at 8,000 xg at RT, the cholesterol present in the HDL fraction (supernatant) was assayed as described above (Randox Laboratories, Crumlin, UK; cat. CH203).
Triglycerides (Tg) concentration was determined after hydrolysis with lipases and spectrophotometrically assessed by monitoring the quinoneimine formation at 500 nm according to the manufacturer’s instructions (Randox Laboratories, Crumlin, UK; cat. TR210).
LDL-Cholesterol (LDL-C) was calculated by the Friedewald equation.24
β-Hydroxybutyrate (β-HB), a ketone body produced by liver from fatty acids, was determined by a colorimetric kit (Cayman Chemical, Ann Arbor, MI USA). β-Hydroxybutyrate dehydrogenase oxidizes serum β-HB to acetoacetate in the presence of NAD+, forming NADH. In the presence of diaphorase, NADH reacts with a colorimetric detector (WST-1) to produce a formazan dye which is detectable at 450 nm.
Statistical analysis
Unpaired Student’s t-test (for normal variables) or Mann-Whitney U test (for non-normal variables) were used to identify significant differences within chronological aging. Significant differences between groups recapitulating endocrine aging were evaluated by ANOVA and Bonferroni post-hoc test or by Kruskal-Wallis followed by Mann-Whitney U test corrected for multiple comparisons, for normally or not-normally distributed variables, respectively. All analyses were performed using SPSS 17.0 for Windows (SPSS Inc., Chicago, IL USA) and GraphPad Prism (GraphPad Software, San Diego, CA USA). A p<0.05 was considered statistically significant.
Results
Expression of lipoprotein and cholesterol metabolism-related genes in rat cortex
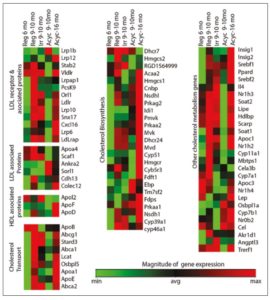
RNA expression levels of 84 genes involved in lipoprotein signaling and cholesterol metabolism were determined in cortex collected from regular 6 month old (mo), regular 9-10 mo, irregular 9-10 mo, acyclic 9-10 mo and acyclic 16 mo rats (Table 1). Differences in gene expression were analysed by ANOVA followed by pairwise t-tests for each transition period (Figure 1).
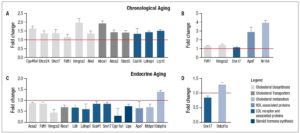
To evaluate the effect of chronological aging before the perimenopausal transition, we compared gene expression profiles of regular 9-10 mo to regular 6 mo rats. As shown in Figure 2A, regular 9-10 mo rats showed a significant up-regulation of genes involved in the synthesis of cholesterol (Cyp46a1, Dhcr24, Dhcr7, Fdft1, Mvd), ketogenesis (Hmgcs2), LDL receptor and LDL associated proteins (Cxcl16, LDLrap1, Lrp10), and cholesterol transporters (Abca1, Abca2, Stard3). Chronological aging after menopause, evaluated by comparing acyclic 16 mo to acyclic 9-10 mo rats (Figure 2B), was also associated with up-regulation of several genes, although fewer genes were significantly altered in comparison to pre-menopause chronological aging. In particular, significant changes were observed only in genes coding for HDL composition (ApoF), LDL internalization (Snx17), ketogenesis (Hmgcs2), cholesterol synthesis (Fdft1), and cholesterol metabolism (Nr1h4).
To distinguish the impact of endocrine aging from chronological aging, we compared regular, irregular, and acyclic rats at the same age (9-10 mo). Irregular cyclers did not show any significant difference when compared with the age-matched regular group (data not shown). We observed a general down-regulation of genes in the acyclic 9-10 mo rats compared with the regular cycle group with the exception of Osbpl1, which was up-regulated (Figure 2C). Significant down-regulation was observed for genes involved in cholesterol transport (ABCA1), cholesterol biosynthesis (Acaa2, Fdft1), cholesterol metabolism (Mbtps1), HDL associated protein (ApoF) and LDL receptor (Ldlr, Ldlrap1, Scarf1, SNx17), steroid hormone synthesis (Cyp11a1, Lipe), and ketogenesis (Hmgcs2). The acyclic group as compared to the irregular group showed significant differences for only two genes, with higher expression of genes encoding oxysterol binding protein (Osbpl1a) and lower expression of LDL receptor associated protein (Snx17) (Figure 2D).
Network and pathway analyses identified gene nodes and upstream regulators during chronological and endocrine aging.
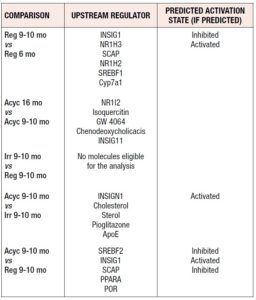
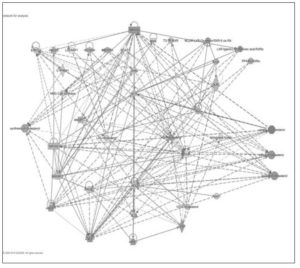
We applied Ingenuity® Pathway Analysis to analyze networks of genes and their upstream regulators in rat cortex. IPA identified thirteen networks related to lipid metabolism during chronological and endocrine transitions, with interactions between genes identified in our dataset and predicted interactions from the IPA knowledge base identified (Table 2). The upstream regulators for each transition are ranked and listed in Table 3 with their activation state (if predicted).
The first ranked network (Figure 3), consisting of 23 foci and 12 interconnected molecules (genes that were not assayed by the array), was subjected to “Function and Diseases” analysis by IPA, focusing on the biosynthesis, uptake, efflux, and export of cholesterol. The predicted activity (e.g. activation or inhibition) of these pathways for each chronological and endocrine transition is summarized in Table 4. Cholesterol metabolism is predicted to be differentially regulated by chronological and endocrine transitions: while pre- and post- menopausal aging showed activation of cortical cholesterol synthesis, export and efflux, the endocrine transitions were associated with an increase in cholesterol uptake and decreases in cholesterol synthesis, efflux, and export (Table 4).
Changes in expression of proteins involved in cholesterol and lipid metabolism
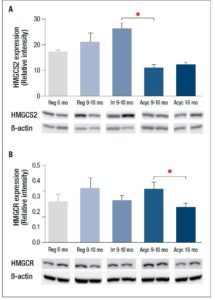
The levels of proteins involved in cholesterol and lipid metabolism were determined by Western blot. The levels of HMGCS2, which forms the substrate for HMGCR, exhibited a significant decrease with endocrine aging (Figure 4A, p<0.01, Acyc 9-10 mo vs. Irr 9-10 mo). The levels of HMGCR, the rate limiting enzyme in cholesterol synthesis, decreased only during post-menopausal chronological aging (Figure 4B, p=0.045, Acyc 16 mo vs. Acyc 9-10 mo), although mRNA levels of Hmgcr showed a non-significant increased trend of expression. Taken together, the protein expression data and the bioinformatics prediction from the gene array suggest that the endocrine transition is accompanied by down-regulation of cholesterol synthesis. Moreover, during pre-menopausal chronological aging, Hmgcs2 exhibited increased gene expression (Figure 2A) and both HMGCS2 and HMGCR showed trends of increased protein expression (Figure 4), consistent with the prediction of cholesterol synthesis activation during endocrine aging (Table 4). However, the IPA predicted activation of cholesterol synthesis was not supported during postmenopausal chronological aging, as HMGCS2 protein levels remained constant and HMGCR protein levels decreased.
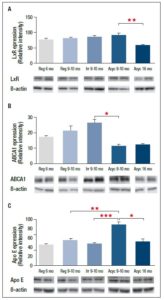
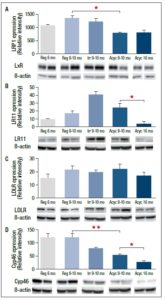
To investigate cholesterol efflux and mobilization in rat cortex, we determined protein expression of the LXR receptor and two downstream proteins: ABCA1, which is involved in cholesterol efflux, and ApoE, which plays an important role in cholesterol transport. LXR showed decreased protein expression during post-menopausal chronological aging (Figure 5A, Acyc 16 mo vs Acyc 9-10 mo), without any difference during endocrine transitions. ABCA1 levels decreased during the endocrine transition (Figure 5B, Acyc 9-10 mo vs Irr 9-10 mo), after a trend of increasing during chronological aging. The protein level of ApoE first increased with onset of acyclicity (Figure 5C, Acyc 9-10 mo vs Reg and Irr 9-10 mo) and then, like LXR, decreased during postmenopausal chronological aging (Figure 5C, Acyc 16 mo vs Acyc 9-10 mo), suggesting a reduced efficiency in cholesterol mobilization during post-menopausal aging.
Next, modulation of cellular uptake of cholesterol through ApoE was investigated by analysing three proteins of the LDL receptor family. Consistent with LDL receptor gene expression results, LRP1 protein levels were lower in acyclic 9-10 mo rats than in the age-matched regular group (Figure 6A, Acyc 9-10 mo vs Reg 9-10 mo). ApoE receptor LR11 levels decreased with postmenopausal chronological aging (Figure 6B, Acyc 16 mo vs Acyc 9-10 mo). On the other hand, LDLR levels did not show any significant change at any stage (Figure 6C). In addition, modulation of oxysterol production as a possible cholesterol clearance pathway was investigated by evaluating protein expression of cholesterol 24-hydroxylase (Cyp46). Cyp46 catalyses the synthesis of 24-hydroxycholesterol (24S-OH-C), the only brain-derived cholesterol metabolite present in the systemic circulation. The levels of Cyp46 protein decreased both during the endocrine transition and during post-menopausal chronological aging (Figure 6D, Acyc 9-10 mo vs Reg 9-10 mo and Acyc 16 mo vs Acyc 9-10 mo, respectively).
Serum lipid profile in rats
As shown in Table 5, serum lipid-related parameters (total cholesterol, triglycerides, HDL, LDL, and ketone bodies), were not significantly different during endocrine transitions (Reg, Irr, or Acyc at 9-10 mo). Conversely, chronological aging was significantly associated with alterations in several lipid parameters. In particular, pre-menopausal chronological aging (Reg 9-10 mo vs Reg 6 mo) was associated with increased levels of total cholesterol and LDL (p<0.05) and a decrease in ketone bodies (p<0.05). Furthermore, during post-menopausal chronological aging (Acyc 16 mo vs Acyc 9-10 mo) there was an increase in triglyceride levels (p<0.05). These findings in a rodent model of female midlife aging are consistent with those derived from midlife aging women below.
Serum lipid profile in women
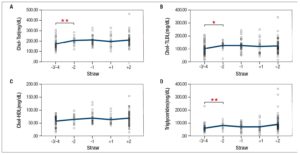
Women were stratified according to STRAW +10 criteria23 into pre-, late pre, peri-, early post-, and post-menopause stages (Table 6). In agreement with this classification, 17-β-estradiol (E2) levels decreased and FSH levels increased during the transition from pre- to peri- and to post-menopause. Anthropometric parameters (BMI, waist, and hip circumference), exhibited a significant increase (p<0.05) during pre-menopausal chronological aging. Endocrine transitions, as suggested by comparisons among early peri-, late peri- and early post-menopause groups at the same age range (Table 6), were not associated with significant variation in lipid profile (Figure 7A-D, -2, -1, +1 groups). Conversely, total cholesterol, LDL-cholesterol and triglyceride concentrations increased during chronological aging prior to menopause (Figure 7A, B, D, -3/-4 vs -2).
Discussion
During the perimenopausal transition, physical and psychological symptoms such as increased body temperature, insomnia, pain, depression and cognitive impairment, are the result of endocrine and chronological senescence.1,2,25 Symptoms are largely neurological in nature and reflect dysregulation of multiple systems, including changes in brain bioenergetics.1,5,6 Indeed, it has been shown that decreased mitochondrial respiration capacity during aging and reproductive senescence in animal models of AD is greater in female mice compared to male mice.5,26 Furthermore, declining glucose metabolism has been shown to coincide with decreased glucose transporters and increased monocarboxylate transporters in aging female mouse brain, with changes appearing earlier in the AD animal model.27 Ketone bodies can be used as a compensatory source of energy in the brain but this leads to catabolism of endogenous structural lipids from white matter and brain atrophy.14,28 Longitudinal brain imaging studies of women during the perimenopausal transition have shown not only that brain glucose metabolism and mitochondrial oxidative capacity decline, but also that brain atrophy and amyloid beta deposition increase with reproductive senescence.11,15,29 These data, along with studies reporting a reduction of cholesterol levels in the cortex and hippocampal areas in AD patients30 prompted us to explore the modulation of lipid metabolism during chronological and endocrine aging. In the present study, we evaluated the modification of brain lipid homeostasis in a rat model that resembles the chronological and endocrine aging of the human perimenopausal transition.1,31 In our rat model, chronological aging was characterized by up-regulation of genes in cholesterol synthesis pathways and those encoding HDL and LDL transporters and binding proteins. These age-related modifications were most apparent during pre-menopause. Conversely, endocrine aging was associated with an overall down-regulation of genes involved in cholesterol metabolism pathways as well as protein levels of HMGCS2, which functions at the branch point of the cholesterol synthesis and ketogenesis pathways. In contrast, a significant reduction in the protein level of HMGCR, the rate limiting enzyme for cholesterol synthesis, occurred later, during post-menopausal chronological aging.
The mechanism underlying activation of cholesterol transport, metabolism and efflux involves the transcription factors liver X receptor (LXR), which can be activated by 24(S)-hydroxycholesterol (24-S-OH-C).22,32,33 In our study, Cyp-46, the primary enzyme involved in the conversion of cholesterol to 24S-OH-C in the CNS, showed a significant decrease in the cortex during both endocrine and post-menopausal chronological aging. 24S-OH-C is the only brain-derived cholesterol metabolite present in the systemic circulation and its serum levels have been correlated to Alzheimer’s disease.34,35 Lower 24S-OH-C levels could lead to decreased stimulation of the LXR pathway, in agreement with the decreased levels of LXR protein we observed in the cortex of aged post-menopausal rats. Our data show that ApoE protein expression increased during the menopausal transition and then decreased during postmenopausal chronological aging. We hypothesize that this decrease may be a consequence of downregulation of the LXR pathway which would impact transcription of ApoE. On the other hand, the significant reduction in ABCA1 observed during the endocrine transition suggests that the decline in ABCA1 might not be connected to the LXR pathway itself. However, the reduction of ABCA1 from perimenopause to menopause is consistent with results found in a mouse model of AD where association between ABCA1 deletion and increased Aβ deposition was observed.36 Furthermore, ABCA1 knock-out mice exhibited reduced ApoE and cholesterol levels in neuronal and glial cells compared with wild-type mice.37 Defects in cholesterol metabolism during the endocrine transition are also associated with a significant drop in LRP1 protein. The reduction of this essential ApoE receptor might impair cholesterol delivery to neurons and affect Aβ clearance across the BBB. Significantly lower levels of LRP1 and Cyp-46, suggesting a progressive accumulation of cholesterol in brain tissue, are consistent with previous histological evidence showing inverse association between the Cyp-46 enzyme activity and Aβ deposition in the medial temporal lobe.38
Finally, considering the brain as part of a “larger integrated biological system”,25 we characterized the systemic lipid profile in both our perimenopause rat model and in a cohort of perimenopausal women. Although metabolism of cholesterol in the brain is different from that of the rest of the body, the two pathways may influence each other as it is known that modifications in peripheral lipid composition can alter oxygen and nutrient supply to the brain.39
Furthermore, 24-OH-C and 27-OH-C derived from cholesterol metabolism in CNS and periphery, respectively, can cross the BBB and act as signaling molecules.40 Alterations in lipid profile are well-recognized risk factors for atherosclerosis and stroke, which contribute to AD risk.37,41 Our data reveal that only chronological aging is associated with a worsened lipid profile, as demonstrated by the increase in total cholesterol, LDL-C and triglycerides during pre-menopausal chronological aging. We note that differences in sample size between groups of women in our cohort may have reduced the power of statistical analysis. Further experiments are required to clarify the relationships between these two systems in humans. In conclusion, the present study advances our previous findings that perimenopause is a transition period characterized by two aging programs: endocrine and chronological. Our data support a defect in cholesterol export during endocrine aging and a subsequent drop in cholesterol mobilization during post-menopausal chronological aging. The decline in cholesterol homeostatic, transport and efflux mechanisms early in the aging female brain is consistent with an early prodromal state and increased risk of Alzheimer's disease later in life.
Acknowledgments
The authors would like to thank the International Society of Gynecological Endocrinology (ISGE) for promoting this work with the best poster award at the ISGE Congress, 2016. This work was supported by University of Ferrara, “contributo 5 per mille - dichiarazione dei redditi 2012” and by NIH/NIA P01AG026572 to RDB: Project 1 to RDB and FY and Analytic Core to FY.
Conflict of Interest
The authors declare that there is no conflict of interest