Introduction
Polycystic ovary syndrome (PCOS) is a frequent disease affecting 4-25% of women of reproductive age depending on ethnicity [1,2]. The diagnostic criteria for this syndrome were established by a consensus meeting in Rotterdam [3]. The diagnosis of PCOS requires that at least two of the following criteria be met: [4] chronic anovulation disorder (oligo or anovulation up to amenorrhea); [5] clinical (acne, hirsutism) or biochemical signs of hyperandrogenism; and [6] presence of micropolycystic ovaries at ultrasound or presence of 12 or more follicles with a diameter of 2-9 mm in each ovary, and/or increased ovarian volume (> 10 ml) [7].
With the evolution of research into PCOS bringing new insights into the syndrome, a new clinical finding has recently been shown to have a high frequency of occurrence in PCOS, namely the dysmetabolic feature of insulin resistance (IR) and correlated compensatory hyperinsulinemia [8,9]. These dysmetabolic situations occur when tissues, due to various modalities, are less sensitive to the presence of glucose in the circulation and therefore require a higher insulin signal in order to be able to take up glucose into the cells. There might be various genetic and/or epigenetic conditions predisposing to compensatory hyperinsulinemia.
Increasing evidence shows a familial aggregation of women with PCOS, hyperandrogenism and metabolic alterations [10]. As yet, there is no well-defined theory that might explain this; while autosomal dominant transmission linked to a single genetic defect has been proposed, most authors define PCOS as a polygenic pathology. The main candidate genes are those encoding factors involved in the synthesis, transport, regulation and effects of androgens, but also genes encoding factors involved in insulin metabolism (i.e. insulin receptors) or in excessive serine phosphorylation of the insulin receptor itself [11,12], signaling cascade proteins responsible for binding of insulin to its receptor, the IGF system, and other growth factors, and finally the gene encoding for Calpain-10 enzyme, responsible for insulin secretion and action [10]. An association has also been found between “proinflammatory” genotypes and PCOS, linked to polymorphism of genes encoding for TNF-alpha, IL-6 and IL6 receptor [10]. However, only a few PCOS susceptibility genes have been repeatedly identified, in studies of women with Chinese or European ancestry [13-24]. All such studies clearly suggested a putative role of genetic factors, but probably the real trigger is the combination of these with environmental factors.
Although the prevalence of PCOS is similar in all countries, ethnic factors influence the phenotypic manifestations of the syndrome. The prevalence of PCOS among Caucasian women ranges from 4.7 to 7% [25] and it has been reported that when groups moved from one place to another, they retained their ethnic predisposition to PCOS, hyperinsulinemia and diabetes, as observed, for example, in the United Kingdom in women of Asian origin [25]. These data clearly infer a role for various environmental factors, such as diet, physical activity and lifestyle in general. Since metabolic disorders occur at a higher frequency rate in developing countries, it has been argued that the pathogenetic triggers of the impairments are associated with lifestyle and survival disadvantages [25]. Such populations have easy access to food that, once eaten and metabolized, produces a high amount of glucose, which is important for brain function but increases IR and induces overweight and/or obesity. Fat accumulation is good to overcome reduced food availability but predisposes to a proinflammatory condition with increased secretion of cytokines and inflammatory factors. All these impairments make the subject more resistant and favor survival in the presence of stressors such as reduced availability of food, injury and epidemics [26]. In the absence of stressors, however, as in the case of developed countries, these pathogenic mechanisms predispose to cardiovascular disease and atherosclerosis [26].
The endocrine profile of PCOS patients
PCOS patients usually show a variety of endocrine abnormalities. Classically, they show elevation of LH, of both ovarian and adrenal androgens, and of estrogens (mainly estrone) due to extra glandular conversion from androgens; they also show reduction of sex hormone-binding globulin (SHBG) and, not rarely, raised prolactin. A specific, and new, issue is the possible finding of increased levels of insulin, in the presence of overweight or obesity but also in normal-weight or lean PCOS patients [27,28].
Although the pathogenesis of PCOS is still under investigation [28,29], the real new issue in this syndrome is the presence of hyperinsulinemia and IR at a higher grade than in the normal female population [30]. While it seems clear that impaired gonadotropin secretion, hyperandrogenism and relative excess of adrenal androgens, and reduction of SHBG are the common endocrine features of the syndrome [28,29,31,32], the dysmetabolic features (i.e. IR and compensatory hyperinsulinemia) have been shown to be a main triggering feature for most of the issues of PCOS [30].
Insulin sensitivity is affected directly and indirectly by androgens, since these may directly inhibit peripheral and hepatic insulin action and clearance. In fact, androgens such as testosterone modulate post-binding signaling, reducing the number and efficiency of glucose transport proteins, such as SHBG, as well as the type 4 glucose transporter (GLUT-4), thus inducing IR in women with PCOS. This effect is particularly evident in the most metabolically active tissues such as muscle and fat [33] and obviously, it is more severe in lower-body obese patients since, due to their increased abdominal fat, they show higher free androgen and insulin plasma levels, as well as higher IR than weight-matched controls [34]. In addition, in obese subjects it has been shown that hepatic insulin excretion and insulin-stimulated glucose uptake in skeletal muscle is improved by free fatty acids and androgens, thus increasing both IR and compensatory hyperinsulinemia [35-37].
In addition, leptin and adiponectin participate as causal factors of IR. Adiponectin, an adipocyte-derived collagen-like protein, is synthesized by adipose tissue and released into the circulation [38,39]. Leptin, another adipocyte hormone, is encoded by the “ob” gene and transmits metabolic signals to the neuronal networks in the brain, where it modulates hypothalamic activity, and also affects the pituitary-ovarian axis [40]. Since PCOS patients, particularly if obese, show reduced adiponectin and elevated leptin plasma levels, a positive correlation has been demonstrated between serum leptin and adiponectin levels and the clinical and hormonal indexes of IR [41]. Moreover, leptin is also linked to neuropeptide Y (NPY) modulation on the reproductive axis thus are both involved in the abnormal reproductive function [42].
Insulin resistance and compensatory hyperinsulinemia
It is clear that IR does not occur due to overweight or obesity per se since the IR observed in obese PCOS has also been observed in normal-weight or lean patients, thus raising the hypothesis of a post-receptor defect that could affect glucose uptake [43,44] other than to be related to excessive serine phosphorylation of the insulin receptor [11,12]. That said, the presence of overweight or obesity worsens IR and the associated compensatory hyperinsulinemia becomes a potent negative modulator of ovarian function, since hyperinsulinemia can increase ovarian androgen synthesis [45-47] and also cause an excess of LH secretion [48,49]. These hypotheses were recently supported by a meta-analysis published by Behboudi-Gandevani et al. [50].
Since PCOS patients can develop abnormal glucose control, frequently evolving into mainly type II diabetes, and this evolution is more rapid in PCOS patients than in normal controls [51,52], screening of women with PCOS for glucose intolerance and hyperinsulinemia is an important clinical step that needs to be performed using at least a 2-hour oral glucose tolerance test (OGTT) [53-55]. It has been clearly shown that any kind of improvement to everyday lifestyle and/or the use of specific compounds (such as metformin) can delay or block both the risk of type II diabetes development and cardiovascular risks [28,53,56].
The fact that hyperinsulinemia has been reported also in non-obese PCOS patients clearly strengthens the hypothesis that excess of insulin secretion is not dependent only on excess of body weight and/or on reduced glucose tolerance [47]. Whatever the event inducing IR and compensatory hyperinsulinemia, it creates the conditions for exaggerated beta cell function of the Langerhans cells in the pancreas and stresses its function thus creating the conditions that, over time, will lead it to exhaust its normal activity, predisposing to cell dysfunction and to diabetes [47]. Furthermore, excess of plasma insulin amplifies, through a specific action on hypothalamic kisspeptin-secretin-neurons, both LH release and the LH-induced production of androgens by the theca cells thus exacerbating hyperandrogenism and hyperandrogenic symptoms in hyperinsulinemic subjects with PCOS [30, 57].
While assessment of compensatory hyperinsulinemia in fasting conditions is important, performing the OGTTto check the insulin response to the glucose load is certainly the optimal approach. The insulin maximal response usually occurs within between 30-60 and 90 minutes after the glucose load and it is considered within normal limits if it is below 50 µU/ml [58]. IR and insulin sensitivity can also be computed by simply calculating the glucose-to-insulin ratio [58,59], whose value should be higher than 4.5, but HOMA index is actually considered more precise and can be computed as the HOMA-IR as follows: (fasting insulin mU/l)×[fasting glucose mmol/l)/22.5 [4].
Hyperinsulinemia is present when the HOMA value is below 2.71 in adults [4,60] and 2.5 in children and adolescents [60]. Recently two different groups suggested that all patients showing fasting insulin plasma levels above 12-13 µU/ml should be considered at risk of compensatory hyperinsulinemia [5,61].
Hyperinsulinemia and dysmetabolism of PCOS
It is current evidence that hyperinsulinemia is central to the pathogenesis of PCOS in many cases, since excess of insulin can induce higher ovarian androgen production and anovulation [62,26], sustained also by the abnormal LH secretion. The frequency of menstrual abnormalities is higher in hyperinsulinemic than in normoinsulinemic women with PCOS [11,12]. IR and compensatory hyperinsulinemia are easily observable in a high percentage of PCOS patients and these are the patients that have an increased risk of developing type 2 diabetes and coronary heart disease (CHD) [63-65]. This risk has also been demonstrated to be higher in postmenopausal women who suffered from PCOS during their fertile life [66,67].
The chance of developing metabolic syndrome (MS) has been reported to be higher in PCOS, since the characteristics of the syndrome, i.e. hyperinsulinemia, mild-to-severe glucose intolerance, dyslipidemia and hypertension, are all risk factors for cardiovascular disease (CVD) and diabetes [68-73]. A significantly higher odds ratio for the development of various cardiovascular risk factors has been reported in PCOS patients compared with the normal population [73].
MS occurs frequently in PCOS (up to 40-45%) [74] and the main predictor factors are hyperinsulinemia, and high free testosterone and low SHBG serum levels [75,76]. The association of MS with PCOS appears to be particularly strong in PCOS women who are young (<30 years) and overweight or obese (BMI > 27 kg/m2) [73].
Women with PCOS have lower HDL levels, higher LDL:HDL ratios and higher triglyceride levels than healthy eumenorrheic women [64]. All these features are inductors of subclinical atherosclerosis, as shown by the increased thickness of the carotid intima media and higher endothelial dysfunction observed in PCOS patients [77,78], probably related to IR and/or to the higher free testosterone plasma levels [79,80]. On this basis it is clear that PCOS patients have a real increased risk factor profile for CVD, and decreased cardiac systolic flow velocity, diastolic dysfunction, increased vascular stiffness, low-grade chronic inflammation, increased oxidative stress, and altered hemostasis, including impaired fibrinolysis and increased tissue plasminogen activator antigen, have, in fact, all been reported in this population [66,81]. In addition, postmenopausal women with a premenopausal history of irregular menses and hyperandrogenism (i.e., PCOS) have more angiographic coronary artery disease than non-PCOS women [66,81], thus providing evidence that identification and treatment of PCOS may represent an opportunity to reduce CVD risk factors [81].
It is clear that PCOS patients who develop hyperinsulinemia have a higher risk of showing a change in glucose tolerance that might lead them towards type 2 diabetes, and that the abdominal phenotype represents an important additional risk factor for women with PCOS [43,66,82,83].
Hyperinsulinemia and PCOS vulnerability
According to what has been reported thus far, IR is a specific biological adaptation that induces compensatory hyperinsulinemia in approximately 70% of women with PCOS and overweight or central obesity; it also occurs in 15-30% of lean women diagnosed with PCOS [3,28] thus reinforcing the concept of a built-in predisposition to a metabolic impairment [27].
On this basis, insulin sensitizers have been proposed as an optimal treatment to counteract this compensatory hyperinsulinemia, together with clear lifestyle changes [28,84]. The best drug currently proposed is metformin, used at variable dosages depending on the severity of the glucose metabolic impairment [28,59,85]. Although safe at high dosages, such as 1.5-2 g or higher, metformin can induce various gastrointestinal side effects such as diarrhea, vomiting and nausea that reduce compliance [86].
Recently a new integrative approach to IR in PCOS has been proposed using inositols in two clinically active isoforms, i.e., myo-inositol (MYO) and d-chiro inositol (DCI) [27], and alpha lipoic acid (ALA) [87,88]. Both inositols and ALA have been reported to be effective. Let us take a look at them in more depth.
Inositols: myo-inositol and d-chiro inositol
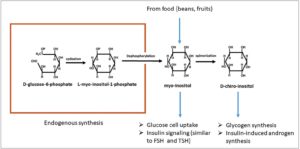
Although inositols first appeared in the clinical approach to IR in PCOS only in the last decade, their story actually goes back a long way. It begins in 1850, when Johannes Scherer isolated, from muscle cells, a new compound that he named “inositol” from the combination of different Greek stems [89,90]. Chemically, inositol is a hexahydroxycyclohexane that formally belongs to the sugar family [91]. Inositols are a family of 9 stereoisomers and the most important, biologically, is MYO since it has been identified as the most widely distributed, being found in all biological systems. For this reason, it has been thought to be a specific probiotic molecule [92]. Inositols are generally found in many plants and in various foods, especially beans and fruit, in the form of derivatives of inositol, that is as hexaphosphate and phytic acid or its salt derivatives (phytates) [91]. Small amounts of inositols are produced also in human tissues [91] directly from D-glucose-6-phosphate through a cyclase that forms L-myo-inositol-1-phosphate, after which through dephosphorylation (inositol monophosphate enzyme) [93], free MYO is constituted [94] (Fig. 1). However, endogenous production is not sufficient for our biological needs, meaning that adequate intake with food is important. Chemically speaking, MYO, although very close to the glucose molecule, belongs to the vitamin complex but it cannot be considered a real nutrient since it can be synthesized by the human body.
Once MYO enters the cell, it is immediately transformed inside cell membranes into phosphatidyl-myo-inositol, which is the precursor of the inositol-triphosphate that is the intracellular second messenger for insulin as well as for FSH and TSH [47,89,95].
At this point it has to be noted that MYO is not the only inositol that serves as a second messenger. In fact, among the other 8 isomers belonging to the family, Larner et al. [94,96] reported that DCI, through different mechanisms compared with MYO, also played an important role in the intracellular transmission of metabolic insulin signals [94,96]. Both MYO and DCI have the same chemical structure but differ in the position of a hydroxyl group. In vivo DCI is synthesized through the activity of an epimerase that converts MYO into DCI. Knowledge of the existence of this epimerase is a key factor for next clinical considerations [30].
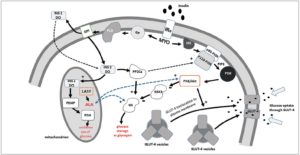
Figure 2 shows a model of the way in which MYO and DCI might, hypothetically, work in the transduction of the metabolic signal of insulin [94,97] (Fig. 2). In brief, the insulin binding to its own receptor activates an insulin receptor (IRe) tyrosine kinase that autophosphorylates. This event leads to the recruitment of insulin receptor substrate (IRS) proteins and phosphorylates these on Tyr residues to serve as scaffolds [94]. A principal IRe/IRS target is phosphatidylinositol-3-kinase (PI3K), which generates phosphatidylinositol (3, 4, 5)-trisphosphate (PIP3) to activate the phosphorylation of a protein kinase (PKB)/Akt by the phosphoinositide-dependent kinase (PDK). After some steps, Akt activation leads to translocation of GLUT-4 vesicles to the plasma membrane to transport glucose into the cells [97].
This is the first pathway. Now we come to the the second, additional pathway, namely the DCI one [97] (Fig. 2). Insulin binding on the receptor activates a G protein (Gq) that is coupled to a GPI phospholipase (GPI phospholipase D, PLD). Activation of the phospholipase produces an inositol glycan second messenger called INS-2 (insulin second messenger with a 4-O-(2-amino-2-deoxy-beta-D-galactopyranosyl)-3-O-methyl-D-chiro-inositol structure) from a GPI lipid precursor in the inner and/or outer surfaces of the cell membrane [97]. INS-2 is then released inside the cytoplasm as well as outside the cell where it can be used by neighboring cells or can re-enter the original cell via an ATP-dependent inositol glycan transporter. Inside the cell, INS-2 activates cytosolic PP2Ca (phosphoprotein phosphatase 2C alpha) and mitochondrial pyruvate dehydrogenase phosphatase (PDHP). In the cytosol, activated PP2Ca stimulates glycogen synthase (GS) directly and indirectly via a PI3K/PDK/Akt/GSK3 pathway (this is the pathway previously described). In the mitochondria PDHP induces PDH and this, in turn, promotes glucose oxidative use. In the cytosol the activation of PKB/Akt leads to inactivation of glycogen synthase kinase 3, resulting in glycogen storage. The activated Akt drives activation of mTOR kinase and then the translocation of GLUT-4 to the plasma membrane [97].
This complicated and interactive model reflects the fact that a specific equilibrium is needed between these two isomers, MYO and DCI, to ensure the best control of the insulin signal. In addition, their presence inside the cell is important, as is adequate intake with food and an adequate MYO-to-DCI conversion through epimerase so as to obtain correct oxidative use of glucose and/or its storage as glycogen. It clear that the presence of MYO is able to activate, through the GLUT-4 vesicles, uptake of glucose. This event reduces glycemia at the intercellular spaces, thus inducing a systemic insulin decrease. On the other hand the role of DCI is to act as a promoter of glucose storage as glycogen and to induce its oxidative use to produce energy at mitochondrion level. These are important activities that improve the reduction of the amount of intracytoplasmic glucose, creating a specific glucose gradient that facilitates additional transportation of glucose through the GLUT-4 vesicles expressed and translocated at cell membrane level.
Alpha lipoic acid (ALA)
Alpha lipoic acid (ALA) is a potent antioxidant that plays a central role in establishing and maintaining the antioxidant defence network by effectively scavenging reactive oxygen species and regenerating critical antioxidants [98,99]. ALA is synthesized inside the mitochondrion and is an essential cofactor of mitochondrial enzyme complexes involved in oxidative metabolism. Endogenously, lipoic acid is synthesized from octanoic acid by the action of lipoic acid synthase (LASY) inside the mitochondrion (Fig. 2). The discovery that mammalian cells are capable of synthesizing lipoic acid, mainly in mitochondria, was made quite recently in the mouse [100]. Although the pharmacological effects of lipoic acid have been explored in many studies, the importance of endogenous lipoic acid is not completely clear. Lipoic acid administration, by virtue of its antioxidant effect, has been found to be beneficial in many metabolic and vascular diseases [101-105] and it has been demonstrated to act on glucose metabolism. Indeed, endogenous ALA modulates glucose utilization through the increase of adenosine monophosphate activated protein kinase (AMPK) in skeletal muscles [4], thus increasing GLUT-4 [106,107]. It becomes clear that both ALA and MYO, independently, play a crucial role in the last and final pathway that activates the GLUT-4 vesicles on the cell membrane that allow the glucose uptake and its entry into the cytosol (Fig. 2).
Impaired metabolism in diabetes and PCOS
The fact that inositol has been considered as a putative integrative treatment for diabetes and PCOS rests on the fact that in both these diseases the metabolic signal of insulin does not work properly. MYO has its own balance with the other eight isoforms, and when requested by metabolic pathways, it is transformed into DCI by an epimerase, with each tissue having its own typical conversion rate [44]. Various studies have demonstrated [108,109] that urinary excretion of DCI is reduced, and urinary MYO is increased, both in humans and in experimental animals affected by type 2 diabetes, and that this situation was not due to the diabetic condition but to an impairment inducing IR [108,109]. The epimerase that determines the conversion of MYO to DCI is insulin dependent, and when IR occurs the amount of DCI production is reduced in insulin-sensitive tissues/organs like the kidney, liver and muscle of experimental animals [110,111]. A marked decrease of epimerase bioactivity was demonstrated in these animal models [112] thus supporting the hypothesis that IR per se is triggered, at least in part, by this kind of impaired epimerase expression.
When diabetic patients were evaluated, lower amounts of DCI and higher concentrations of MYO were observed not only in tissues but also in the urine in baseline conditions [94], and MYO amounts were further increased in comparison to non-diabetic controls after insulin administration [108]. This DCI deficit was expressed as a MYO-to-DCI ratio, which was found to be higher not only in type 1 and type 2 diabetes patients but also in non-diabetic relatives of diabetic patients [94,139]. All these facts support the hypothesis that the diabetic condition and familial predisposition to diabetes induce an abnormal function/expression of epimerase activity, triggering the systemic occurrence of IR and the associated compensatory hyperinsulinemia. When PCOS patients were evaluated for their urine excretion, it was demonstrated also a lower amount of DCI in blood than control subjects with no difference in MYO. In addition, PCOS patients undergoing a glucose tolerance test showed a three-fold lower release of the chiro-inositol glycan (DCI) than control subjects [113] did. Moreover, the IR observed in PCOS patients predisposes to a greater likelihood of developing type 2 diabetes, especially when the patient has overweight/obesity and there is a familial predisposition to diabetes (i.e. diabetic relatives in the family) [114].
To better understand the epimerase story, we have to focus a little more on the fact described up to now. To function perfectly, the inositol system relies on an adequate equilibrium between MYO and DCI. Indeed once insulin binds to its receptor, MYO is specifically activated on the other side of the cell membrane and, as described above, it has two pathways it can follow: one leads it through several steps to induce AMPK activation, and thus to induce glucose uptake through the GLUT-4 vesicles, the other one takes MYO towards its transformation, thanks to the action of epimerase, into DCI, and through the action of DCI two events take place: increased storage of glucose as glycogen and increased oxidative use of glucose [97]. These two latter events are fundamental since they both eliminate glucose from the cytosol and create a concentration gradient that further enhances the upltake of glucose into the cell.
Nevertheless, the impaired epimerase activity is not probably the only impairment. Indeed, studies have demonstrated that type 2 diabetes downregulates the expression of LASY, which is responsible for the synthesis of ALA inside the mitochondria of mammalians [98,100]. In fact, reduced endogenous ALA synthesis induces a decrease in mitochondrial lipoic acid availability; this induces lower glucose uptake in skeletal muscle cells that, in turn, participates in the triggering of IR [98].
Integrative use of inositols and ALA
A large body of data has been published on the administration of inositols in PCOS patients, with all demonstrating that integrative use of inositols was effective in improving insulin sensitivity and restoring menstrual cyclicity and ovulation. Recently, Unfer et al. [115] reviewed the clinical outcomes of MYO administration in PCOS patients, to improve both metabolic and hormonal parameters. Although the clinical use of MYO was not found to be particularly homogenous, since the daily dose used varied from 500 to 1500 mg, the authors stated that PCOS patients showed significant improvements in most of their reproductive hormonal parameters, as well as improved lipid profile, insulinemia and HOMA index [115]. BMI and menstrual function were also improved, as was fertility. This overview supported the hypothesis that the reduction of insulinemia induced by MYO was based on the increased bioavailability of an IPG (Inositol Phospho Glycan) insulin second messenger and that once the endocellular metabolic system works better, endocrine function as a whole starts to work properly again [115].
Though such considerations are relevant, recent data suggest that probably not all is as it seems. In fact, even though MYO is able to improve insulin sensitivity and most hormonal parameters in obese PCOS [5], Genazzani et al. demonstrated that not all obese PCOS patients showed significant metabolic improvements under MYO administration [116]. The presence of a fasting insulinemia below or above the cut-off of 12 µU/ml seems to be the factor making the difference: PCOS patients with insulin plasma levels below 12 µU/ml, despite reducing their BMI, showed minimal or no metabolic improvement in terms of insulin response to the oral glucose load [116]. Such results suggest that obesity is not the only element triggering the hyperinsulinemia and that a built-in abnormal mechanism, or mechanisms, might probably be responsible for the insulin-resistant condition in most but not all PCOS patients. These observations suggest that in these patients, compensatory hyperinsulinemia is probably not resolved by MYO administration due to reduced DCI-IPG synthesis/release. In fact, Cheang et al. [117] suggested the presence of a functional defect (e.g. intracellular defect in the formation or release of the DCI-IPG mediator) rather than a simple nutritional deficiency of inositol, thus promoting the idea of an impairment of epimerase expression/synthesis.
The fact that epimerase activity/expression might be abnormal in diabetic patients and abnormal/reduced in PCOS patients with familial diabetes [94] suggests that the anamnestic evaluation of each single PCOS patient is fundamental. In fact, when obese PCOS patients were administered DCI at the daily dose of 500 mg, all of them showed significantly improved insulin sensitivity [118,119]. However, when all obese PCOS patients with familial predisposition to diabetes were compared with those who had no such familial predisposition, it turned out that PCOS patients with familial predisposition to diabetes had a greater hyperinsulinemic response to glucose load before treatment (and thus a higher IR) [118] and that DCI administration improved insulin sensitivity similarly to what was seen in the other group (with no familial diabetes). Put simply, this observation shows that predisposition to diabetes is probably linked to reduced epimerase expression/synthesis that reduces endogenous MYO conversion to DCI. Thus, DCI administration in these patients significantly restored the adequate DCI levels, allowing improvement of insulin sensitivity [118]. The presence of familial diabetes predisposes PCOS patients to defective synthesis/formation of DCI and DCI-IPG thus triggering IR and predisposing to diabetes. This observation is in agreement with Cheang et al. [117], who argued that multiple defects in DCI metabolism (low availability of DCI or low endocellular synthesis of DCI and/or DCI-IPG) may determine the same result.
An additional aspect to point out is the fact that treatment with metformin, the most known and used insulin sensitizer, has a good and positive effect on ovarian function as well as on the hormonal pattern, mainly through significant reduction of the androgenic milieu [120]. This positive effect of metformin administration is due to a specific action on the release of DCI-phosphoglycans: it thus supports the view that insulin sensitizers improve insulin sensitivity by acting on inositol-based signaling [121]. Indeed, this explains why, in the presence of abnormal DCI-phosphoglycan release, metformin administration might be less efficient in improving PCOS hormonal and clinical signs [122], and it suggests that only higher doses of metformin might be effective.
Let us try to be objective for a moment: it seems strange to think that biological control of glucose uptake into the cell could rely only on one control system, i.e. the inositol pathway. In fact, a second pathway, independent of the inositol one, has been demonstrated and it is driven by mitochondrial ALA production [98]. As previously discussed, ALA modulates and improves glucose utilization by increasing AMPK in skeletal muscles, and thus increasing GLUT-4 [87,88]. Considering that the inositol pathway, through MYO, acts precisely on AMPK, ALA represents a sort of “security system” able to control the final steps in glucose uptake. On this basis, ALA has actually been considered a sort of pleiotropic compound with potential therapeutic use in diabetes and other endocrine diseases [123,124] such as PCOS [106] to improve dysmetabolic-induced impairment.
Since inositols are for the most part introduced with food, unlike ALA, which is synthesized inside the cells, in in the event of a deficit in inositol intake through food, ALA represents an endogenous and autonomous system that might sustain GLT-4 expression so as to maintain regular uptake of glucose from intercellular spaces and avoid a lack of energy. However, as for any complex and integrated system, ALA has also been shown to be affected by diabetes. In fact, the presence of type 2 diabetes downregulates the expression of LASY which is responsible for ALA synthesis in mammalian mitochondria [98,100]. In fact, reduced LASY expression decreases endogenous ALA synthesis, leading to a decrease in mitochondrial lipoic acid. In diabetic subjects, this situation worsens the already impaired glucose uptake in the cells that is at the basis of IR [98], leading to a reduced action on AMPK in skeletal muscles [98], thus further reducing the action on GLUT-4 expression/synthesis [88]. In fact, when ALA was administered to obese hyperinsulinemic patients, a constant reduction of IR was observed, especially in PCOS cases with familial diabetes [30].
At the end of the story, it is evident that the putative combination of defects in expression/synthesis of epimerase and LASY is an important risk factor for compensatory hyperinsulinemia, abnormal control of glucose metabolism and further risk of diabetes.
Clinical considerations on MYO, DCI and ALA integrative administration in hyperinsulinemic PCOS
Although the integrative approach to PCOS is consistently useful and well worthwhile [125], it is clear that before deciding what to propose, among inositols and/or lipoic acid, an anamnestic evaluation is certainly necessary. This consideration hinges on the fact that the natural history of the patient, together with the familial anamnesis, gives, with a higher degree of probability, an indication with regard to the presumed presence/absence of a significant dysmetabolic impairment. Moreover, compensatory hyperinsulinemia, if found, is a risk factor for future health, during pregnancy as well as during the menopausal transition [126].
Numerous papers have demonstrated that the use of inositols, both MYO and DCI, is beneficial in terms of improving insulin sensitivity [91,97,115,116,118-120,125,127,128], but very few of the many groups working on this topic have stressed the aspect of familial predisposition to diabetes.
Indeed, recent studies [113,129] have clearly reported that urinary excretion of MYO and DCI was altered in the presence of diabetes or if patients had diabetic first-degree relatives. In fact, the MYO-to-DCI ratio is higher in these patients than in control women, which suggests that some kind of abnormal metabolic step, such as a reduced expression/synthesis of the epimerase enzyme, is impairing MYO conversion to DCI.
Since not all obese PCOS patients show a reduction of the hyperinsulinemic response to OGTT when undergoing integrative treatment with MYO [5], simple daily administration of DCI (500 mg) was tested in a group of obese PCOS [118]. DCI administration was found to be effective in improving insulin sensitivity as demonstrated by the significant reduction of the insulin response to the OGTT [118]. In addition, when the patients were subdivided according to the presence or absence of any relatives with diabetes (type 1 or 2), it emerged that those with familial diabetes had a higher insulin response than the other group, and that after the DCI integrative intervention, insulin response was significantly and similarly reduced in both groups [118]. This observation clearly supported the evidence that DCI administration reduces IR, restoring almost normal insulin sensitivity. Since epimerase is suspected of being the “weak point” of the enzymatic cascade that regulates DCI synthesis from MYO, these data suggested that predisposition to diabetes negatively modulates insulin sensitivity in PCOS due to the impaired DCI synthesis.
On this basis, it becomes relevant to understand what role ALA administration plays in the control of insulin sensitivity in obese patients. Again, another group of obese PCOS patients was simply administered ALA at a low dosage (400 mg) every day for 3 months [16]. As expected, all patients showed improved insulin sensitivity with no distinction between the presence or absence of familial diabetes. However, the study reported that the only significant changes observed concerned metabolic parameters such as insulin, C peptide, and HOMA index, with no changes reported in relation to reproductive hormones (i.e. LH, FSH, estradiol). Interestingly, this study disclosed that obese PCOS patients with familial diabetes had ALT and AST values at the higher levels of normality and higher than those recorded in the other group of PCOS patients with no familial diabetes; it also showed that ALA administration reduced these high levels [30]. These latter two parameters are relevant to hepatic functioning and it is important to remember that hyperinsulinemia per se is a predisposing factor for MS [126] as well as for non-alcoholic fatty liver disease and hepatic steatosis [62,130].
What about the effects when combining ALA with inositols? Does the combination give any significant improvement of insulin sensitivity with reduction of the compensatory hyperinsulinemia? Most of the studies on this integrative approach dealt with the reproductive outcomes [106,115,119,127,131] and less attention has been paid to compensatory hyperinsulinemia and to familial diabetes as a predisposing factor. Recently a study was designed to elucidate the effect of MYO or ALA, or a combination of both, on impaired insulin release in obese PCOS [132], evaluating the insulin maximal response to OGTT before and after 12 weeks of integrative treatments. The study demonstrated that the HOMA index was improved in all treatment groups, but MYO was able to reduce the insulin response only in obese PCOS patients without familial diabetes. ALA or ALA+MYO administrations were both able to induce a significant decrease in the insulin response under the OGTT independently of the presence of diabetes [132]. These data are in agreement with what is reported in a group of obese PCOS patients [7], namely that the combination of ALA+MYO affected compensatory hyperinsulinemia, blunting the failure observed for MYO when administered to PCOS patients with familial diabetes [7,132]. This result confirms the key role of ALA in the control of insulin sensitivity, and also the fact that ALA overcomes the enzymatic impairment induced by familial diabetes (i.e. LASY defect). In fact, MYO modulates insulin better in the absence of familial diabetes. Probably it would be relatively more efficient administered at high dosages (2 or 4 g every day), but in obese PCOS patients with familial diabetes the consistent limitation due to an impaired epimerase activity would not have been overcome.
Considering that MYO administration appears to work properly only if epimerase is efficiently expressed [132] and that DCI administration has been demonstrated to work effectively in PCOS in general [119], since DCI administration is able to overcome the problem of impaired epimerase expression (in familial diabetes) [118], it seems evident that we should think about using ALA+DCI in combination. In fact, Cianci et al. [127] reported consistent improvements in reproductive and metabolic outcomes in PCOS patients. More recently, our group reported that the ALA+DCI combination (300 mg and 500 mg, respectively) was able to induce greater improvements in obese PCOS patients with familial diabetes, especially with regard to fasting insulin and lipid profiles. In addition, this study showed a significant reduction not only of the maximal insulin response to the OGTT, but also of C peptide in obese PCOS patients with as opposed to without familial diabetes, with the latter group showing no significant modification [133]. It is also very interesting to note that predisposition to diabetes seems to be linked to higher hepatic vulnerability. Indeed, these patients showed higher plasma ALS and ALT levels (in any case within the upper level of the normal range) that were significantly decreased after the treatment interval [133]. These data support the hypothesis that women with familial diabetes have not only a higher predisposition to develop hyperinsulinemia per se, but may also have a strong chance of having abnormal hepatocyte function that might lead to liver steatosis [62,130,134]. It is worth mentioning that a recent paper described an impaired hepatic insulin extraction (HIE) index in PCOS with hyperinsulinemia that decreased significantly under an integrative treatment with a mix of nutraceutical compounds, but no ALA or inositols; in addition to having a significant effect on metabolic pathways, the treatment also improved liver function [134]. These data clearly suggest that hyperinsulinemia is related to overweight/obesity and/or to a genetic defect (i.e. of epimerase and LASY), but probably also to the simultaneous presence of abnormal hepatocyte function in terms of insulin clearance. In fact, almost all insulin extraction is due to the liver and if this is reduced a higher amount of insulin will remain in the blood flow, reinforcing hyperinsulinemia [134].
To better understand the pathophysiological dynamics we have to remember that after proinsulin is included into vesicles in the Golgi apparatus (beta-granules) of Langherans cells inside the pancreas islets [135], insulin molecule is created after the C-peptide is removed, leaving the A-chain and B-chain bound together by disulfide bonds. It is clear that C peptide reflects the β-cell secretory capacity, since its hepatic extraction, unlike that of insulin, is negligible [136,137]; on the contrary, the liver [137] mainly clears insulin. In terms of molar equivalence, the production ratio between insulin and C peptide is theoretically 1, but the ratio that can be computed between plasma insulin and C peptide concentrations reflects greatly the clearance kinetics of both peptides and represents a clear index between the amount of available insulin in the circulation and its release from the pancreatic cells and activity, the latter represented by C peptide [134]. This ratio cannot be equal to 1 both in physiological and in pathophysiological conditions, as demonstrated when using catheterization of the hepatic artery that enters the liver: at the entrance, the patterns of C-peptide and insulin were qualitatively similar, but not when exiting, since insulin is cleared by the liver [138]. In the typical hyperinsulinemia of obese patients, this pattern changed in the hepatic vein according to the liver extraction of insulin, which is reduced, thus participating in the occurrence of the compensatory hyperinsulinemia [138]. This HIE evaluation showed that no changes were observed in normoinsulinemic obese PCOS, which suggests that not all obese PCOS patients are the same in terms of the dynamics of insulin secretion, sensitivity and degradation [134]. Unfortunately, this study evaluated obese PCOS patients on the basis of the presence or absence of hyperinsulinemia at OGTT and did not consider familial predisposition to diabetes, but according to previous studies, the rate of concomitant presence of both familial diabetes and hyperinsulinemia has been reported to be as high as 87.5% [7].
Taken together, these clinical observations give a clear idea of the complexity of PCOS and the related presence of compensatory hyperinsulinemia. They show that an accurate anamnestic investigation is important to discover any diabetic cases among first-/second-degree relatives (parents and/or grandparents) since this might raise the suspicion of some metabolic impairment.
Concluding remarks
PCOS is certainly “not easy to cure”. Specific expertise is needed to understand whether any predisposing factor is present and a good anamnestic evaluation must always be done.
Integrative use of inositols and ALA is definitely helpful and might be proposed to PCOS patients, especially if overweight or obese, always together with a dietary program that reduces intake of carbohydrates to the minimum amount required, thus reducing the amount of glucose derived from food.
The use of MYO and DCI should be encouraged, bearing in mind that, should there be any suspicion that epimerase is not properly expressed and/or functioning, DCI is the best choice. Obviously, MYO+DCI combinations are useful as well, but are probably more indicated for PCOS with no familial diabetes. In fact, in cases with diabetes in the family, DCI is less produced from MYO and the only amount available is the DCI contained in the integrative administration of MYO+DCI. This amount of DCI is probably below the rate biologically needed. We have to remember that although MYO is needed at a higher intracellular concentration to promote AMPK activity, DCI is just as important since it induces glycogen synthesis and oxidative use of glucose, favoring glucose uptake into the cell.
Though ALA is a perfect controller of insulin sensitivity, the biological equilibrium relies on the presence of both the ALA and the inositol pathways. This is why combinations of MYO or DCI with ALA are of great interest. Their advantage lies in the fact that ALA is normally an independent regulator of glucose uptake through GLUT-4 induction and thus of insulin sensitivity. In cases of PCOS with familial diabetes, that is in presence of reduced LASY (and probably epimerase) expression/function, ALA administration overcomes the problem: it jumps over the obstacle! The best solution seems to be the combination of ALA and inositols (MYO or DCI) that can induce a great improvement in insulin sensitivity in all PCOS patients (with or without familial diabetes), but according to the recent reports, ALA+DCI seems to be more effective not only on the specific control of hyperinsulinemia but also on that of lipid profiles.
Acknowledgments
Prof. Alessandro D. Genazzani thanks the whole team of fellows and doctors working at the Gynecological Endocrinology Center, Department of Obstetrics and Gynecology, University of Modena and Reggio Emilia, Italy. Special thanks to Dr. Giulia Despini and Dr. Alessia Prati for the clinical follow-up of the patients at the Center, and to Dr. Alba Manzo for the text revision.
Conflict of interest
The author declares not to have conflict of interest.