Introduction
Tetrasomy X (48,XXXX) is a very rare condition in women, which was described for the first time by Carr et al. in 1961 [1] and to date about 100 cases have been reported worldwide and up to 60 women have been described in scientific publications [2-5]. The analysis of described cases has shown that the tetrasomy X phenotype is characterized by pronounced variability without specific pathognomonic characteristics, which creates problems in its diagnosis and may lead to underdiagnosed cases [3,6,7]. Of the most frequent aneuploidies of sex chromosomes, only the 45, X karyotype (Turner syndrome) and 47, XXY karyotype (Klinefelter syndrome) are characterized by pronounced phenotypes, whereas 47, XXX and 47,XYY karyotypes are not manifested with specific phenotypes [3,8,9]. In general, polysomes of the X chromosome, with the exception of trisomy X in phenotypic females, are rare in the population, and sexual development disorders are not systematically observed in such individuals [2,10].
The most common reason for karyotyping in cases of tetrasomy X is mental retardation, however, 48,XXXX karyotype is also detected in the examination of patients with primary and secondary amenorrhea or speech disorders [5,11-15]. About 50% of girls with tetrasomy X have normal pubertal development, while the other 50% have delayed puberty, partial puberty, complete puberty with menstrual irregularities and/or premature ovarian insufficiency (POI) [5,16]. Adolescents and adult women with tetrasomy X account for 1/3 of the patients described in the literature, about 50% of them have menstrual irregularities [2,5,16,17]. Out of the described tetrasomy X cases, only few patients have been diagnosed with POI [5,15,18,19]. Considering the small number of tetrasomy X cases described in the literature and the prevalence of children and adolescents among them, it is possible that the actual rate of POI in cases of tetrasomy X is higher.
According to the literature, more or less manifested psycho-motor retardation appears to be the most systematic characteristic of tetrasomy X, which is accompanied by speech disorders [14,20-22]. In individuals with tetrasomy X, the average IQ varies from 60 to 80 [2,12,16,21]. Clinical findings suggest that IQ levels decrease by 10-15 points for each additional X chromosome [2,5]. As an exception, it has been described that women with normal IQ who have severe articulation problems and poor speech skills [13]. Thus, it is currently difficult to determine whether speech disorder is an independent characteristic of tetrasomy X or the result of mental retardation, as suggested by some authors [16]. In cases of tetrasomy X, more or less pronounced, sharply variable behavioral problems are common. Some of such individuals are friendly, pleasant, some are aggressive and emotionally labile [2,11,16]. In some cases of tetrasomy X, schizophrenia, epilepsy and hypersomnia have been described [2,23-25]. Based on the limited clinical data, it is difficult to determine whether these abnormalities are related to karyotype features or are due to coincidence, considering that patients with mental disorders are more often subjected to chromosome analysis than individuals with normal mental health.
In individuals with tetrasomy X, non-specific dysmorphic signs, skeletal and connective tissue abnormalities are manifested. There are frequent cases of epicanthus, hypertelorism, flattened nose, up slanting palpebral fissures, irregular teeth, microgenia, clinodactyly, radioulnar synostosis, and joint hyperlaxity [2,14-16,20,26]. In some patients, vision disorders, pathologies of the cardiovascular and renal systems, among others, have been described [2,17,27,28]. Height-related issues are of particular interest in cases of tetrasomy X. According to the literature, the average height of individuals with trisomy X, tetrasomy X and other forms of polysomy X in general exceeds the average height of individuals with normal karyotypes [2,3,5,8,29]. It is postulated that tall stature of patients with extra X chromosomes are related to the positive dose-dependent effect of the short stature homeobox (SHOX) gene in the pseudoautosomal region of the X chromosome related to height [5,29-32]. Also, tall height in patients with polysomy X may be due to a combination of overdosage of the SHOX gene and estrogen deficiency [5,30,31]. Ottesen et al. [33] found that increased number of sex chromosomes affects height in a nonlinear fashion. Other mechanisms that affect height outcome regardless of overdosage of the corresponding genomic regions have also been discussed [30,32]. Thus, it is clear that the literature describes patients with tetrasomy X as either short, normal, tall, or very tall [5,17,19,33].
Several cases described in the literature indicate that fertility is possible in women with tetrasomy X. One woman with tetrasomy X has two children - one with a normal chromosomal complex and the other with Down syndrome. A second woman has one child with a normal chromosomal complex, and the third woman has a stillbirth child with omphalocele. According to the latest data, another woman has 3 children with normal chromosome complex [16,34].
Mechanisms of formation of tetrasomy X have been studied only in a part of the cases described in the literature [11,15,35]. These studies identified double successive maternal nondisjunction at meiosis as the most common mechanism, although more rarely, X tetrasomy can be the result of meiotic errors in both parents or errors at early mitotic division and also as a result of the maternal trisomy X [11,15,35,36]. Scant literature data on tetrasomy X does not allow determining the role of maternal age in these cases [13,37].
Paucity of cases described in the literature also does not allow identification of specific signs typical for this pathology, including the real frequency of POI in this group of patients. It is very interesting to note that in the recent literature, the description of tetrasomy X cases is rare. Thus, the description of each new tetrasomy X case is very interesting and necessary to clarify the phenotype. We here report the case of POI in an adolescent girl with 48,XXXX karyotype.
Case report
A 17-year-old adolescent girl was referred to the Center for Reproductive Medicine "Universe" (Tbilisi, Georgia) in 2021 due to secondary amenorrhea. She was born by vaginal delivery after a full-term pregnancy. Weight at birth was 3000 gr and length 50 cm. There was asphyxia at birth. Age of parents at conception: mother - 20 years and the father - 26 years. Patient is the first child in the family. She has two younger brothers - 8 years old and 10 years old. All members of the family are physically healthy and of normal mental development.
Since infancy the girl had feeding problems such as regurgitation and vomiting and retardation in physical development. Although the patient underwent examinations and treatments in different clinics, the time of consultation parents could not specify the type of received treatments.
On the basis of treatment by the age of 1 year, episodes of vomiting were no longer observed, nutrition was restored and physical development began to improve. From this period on she could sit, started walking at the age of 1 year and 6 months.Teething started at the age of 1 year and 2 months. From the age of 3 she could say single words, but could not speak.She started to speak at the age of 5 after the involvement of a speech therapist. She was treated according to the prescription of their neurologist from early childhood (specific data could not be obtained). She also underwent physiotherapeutic procedures.
When she was referred to us, she spoke with simple sentences, but had defects in the pronunciation of particular sounds. Among peculiarities of the character, it should be mentioned that she was disobedient, withdrawn, had no friends, and would get easily irritated and angry.
Neurological examination was unremarkable. Patient had moderate learning and intellectual disability. Intelligence test detected an IQ of 59, meaning general cognitive functioning was within mild intellectual impairment and difficulties in planning and regulating cognitive processes werealso revealed.
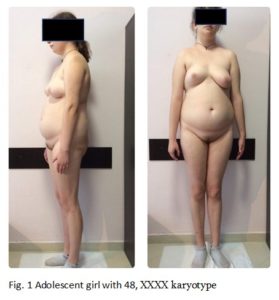
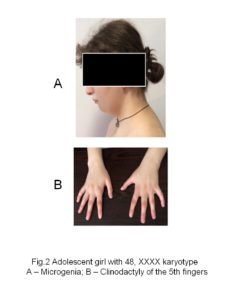
At the age of 17, when consulting to the clinic, objectively she had a height of 177 cm, a body weight of 81 kg (body mass index: 25), and a head circumference of 55 cm (Figure 1). Hersubcutaneous adipose tissue was distributed mainly abdominally, presenting a swollen, abdomen, flat buttocks, microgenia (Figure 2A), high-arched palate, and irregular teeth with poor enamel. The shape her eyes and the shape and placement of the ears were normal, hypertelorism was not observed. Hearing and vision were also normal. Clinodactyly of the 5th finger (Figure 2B) was detected on both upper extremities, more marked on the left. Development of secondary sex characteristics according to Tanner was Ma4, P4, Ax3, areolas of mammae were pale with inverted nipples. According to the gynecological examination: female type of pubic hair, external genitalia and labia majora were normal and properly developed. Labia minora was hypoplastic, yet the clitoris was not virilized.
Regarding menstrual function, she had the menarche at the age of 13, had 3 regular periods, then developed amenorrhea and spontaneous menstruation 1 time per year for 2 years with light menstrual discharge. She has not had menstruation for the last 2 years.
Results of the conducted laboratory examinations upon the visit to the clinic were: FSH 24.2 mIU/mL (Normal 0.3-7.00); LH-23.38 mIU/mL (Normal 0.9-13.3), prolactin 14.37 ng/mL (Normal 4.8-23.3); DHEA 1.21 µmol/L (Normal 0.65-3.68); Estradiol 23.0 pg/mL (Normal 25-345); Total testosterone 0.6 ng/mL (Normal <0.6); TSH 3.8 µIU/mL (Normal 0.51-4.30); FT4-–15.51 pmol/L (Normal 12.60 –21.0); Fasting insulin 57.91 µU/mL (Normal 2.6-25.0); Fasting plasma glucose 108 mg/dL (Normal 60-100); HOMA-IR 15.4 (Normal <2.5); Hemoglobin A1c (HbA1c) 5.5% (Normal 4.2-5.7%); 25-hydroxy vitamin D 12.72 ng/mL (Normal 20-50).
Transabdominal ultrasonographic examination revealed uterine size 19 x 15 x 21 mm, volume 3.1 cm3, cervix and corpus ratio 1/1, endometrial thickness 1.2 mm, right ovary size 14 x 7 x 8 mm, volume 0.4 cm3, follicular apparatus was not visualized, left ovary size 14 x 8 x 9 mm, volume 0.5 cm3, one follicle was fixed in it, size 1.5 mm. Ultrasonography of the thyroid gland detected hypoplasia, volume of the thyroid gland 3.3 cm3 (Normal 14-18 cm3), without structural changes. Breast ultrasonography showed glandular tissue as single fragments with unclearly contoured small-sized structures, excess of fat particles and fibrous tissue.
Brain MRI scan did not detect any significant focal organic pathology. Small venous anomaly was revealed in the right parietal lobe, with 5 mm avascular fluid focus – Rathke’s cleft cyst - seen laterally in the adenohypophysis, Sella Turcica displayed straight, clear contours.
Decrease in bone mineral density - osteopenia - was identified by DXA scanning (Z score –1.3 SD). X-ray test did not detect radioulnar synostosis.
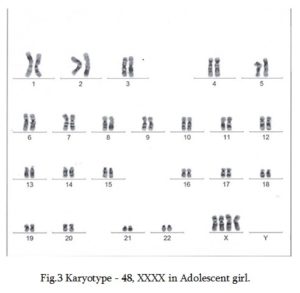
Cytogenetic study of G-banded chromosomes obtained from lymphocyte cell culture at 50 metaphases detected karyotype 48,XXXX (Figure 3).
Based on the conducted examinations the diagnosis was established: X chromosome tetrasomy – 48,XXXX, asymptotic Rathke’s cleft cyst, insulin resistance, hyperinsulinemia, and vitamin D deficiency. The patient was prescribed: Estradiol valerate 2 mg once a day, myo-inositol, vitamin D3 and calciumThe patient was advised about the need for a healthy lifestyle, low-calorie diet, physical activity and dynamic supervision.
After 2 months upon follow-up, the patient lost 5 kg. According to ultrasonography uterine size increased to 24 × 21 × 31 mm, volume - 8.1 cm3, endometrial thickness increased from 1.2 mm to 3.7 mm, size of the ovaries did not change, and follicles were not visualized. The patient stopped treatment and did not show up for the next visit for 8 months due to the COVID-19 pandemic and family problems. At the follow-up visit after 8 months of discontinued treatment, the patient's height was 179 cm, she had grown by 2 cm, and X-rays detected epiphyseal growth plate closure. Densitometry no longer detected osteopenia - bone mineral density corresponded to age-normative indicators.
The patient continues therapy and supervision at our clinic. She is also under the supervision of a neurologist and undergoes speech and physical therapies with relevant specialists.
Discussion
Tetrasomy X-48,XXXX is a very rare condition, characterized by pronounced variability without specific pathognomonic features. POI in patients with tetrasomy X has been described only in few cases [5,15,19]; however, considering that tetrasomy X is a rare pathology and children and individuals in early puberty prevail in described cases, the incidence of POI in this group of patients may actually be higher. It should also be noted that approximately half of puberty age individuals with tetrasomy X experience irregular periods and/or incomplete puberty, which can be further manifested as a POI [2,5,16,17].
Despite a timely menarche (13 years), our patient developed secondary amenorrhea within a few months. Examinations performed at the age of 17, diagnosed POI - hypergonadotropinemia, and ultrasonography showed almost complete depletion of follicles in the ovaries. It is also noteworthy that despite timely menarche and several regular menses, our patient likely had ovarian insufficiency and low estrogen secretion since early puberty, which is indicated by the insufficient expression of glandular tissue in a fragmented form in the mammary glands, pale areolae and underdeveloped nipples with an excess of fatty and fibrous tissue against fairly large mammary glands identified by ultrasonography performed at the age of 17.
According to some researchers, each extra X chromosome affects ovarian function; a higher number of X chromosomes increases the rate of POI and lowers the age of its expression, supposedly due to earlier depletion of ovarian follicles [2,5,15].
It is of great interest to discuss the mechanisms of high stature in patients with tetrasomy X and POI. It is possible that high stature in patients with polysomy X may be due to a combination of SHOX gene overdosage and estrogen deficiency [5,30,31].
Our already tall patient (height 177 cm) gained 2 cm in height (became 179 cm) in the next 8 months since the first visit. During the period of estrogen treatment, the patient stopped growing, and growth zones were found to be closed. It should be assumed that since early puberty, hypoestrogenemia contributed to the patient's growth in height and inhibited ossification of growth zones.
Our patient had psycho-motor retardation (IQ-59) and speech defects, which are most characteristic of tetrasomy X and is actually systematic in similar patients described in the literature [20-22]. Interestingly, she had feeding difficulties before the age of 1. Feeding difficulties in early childhood have also been described in several other tetrasomy X cases [4,38]; however, at this stage it is difficult to tie these difficulties to the peculiarity of the karyotype.
Given the paucity of literature data it is also difficult to determine the association of Rathke's cleft cyst with polysomy X. By means of an MRI a small asymptomatic Rathke’s cleft cyst was diagnosed in our tetrasomy X patient. In only one case described in the literature patient with 48, XXXX karyotype had Rathke’s cleft cyst, however, in this case, a combined multiple pituitary hormone deficiency was detected, supposedly by exerting pressure on the pituitary gland in the sella [39].
Heterogeneous behavioral disorders are characteristic for patients with tetrasomy X [2,11,16]. Our patient was withdrawn, but occasionally aggressive. In cases of tetrasomy X, the presence of non-specific minor physical anomalies poses particular diagnostic difficulties [14,15,20,26]. From a wide range of these minor anomalies described in the literature, only microgenia, high-arched palate, clinodactyly of the 5th finger of both upper extremities were detected in our patient.
The genesis of X polysomy due to trisomy of the maternal X chromosome was excluded, by examining the karyotype of the patient's mother and determining karyotype 46,XX for normal women. X polysomes in females are manifested by wide variability of non-specific phenotypes, which increases the possibility of underdiagnosed cases, however, apart from trisomy X, other X polysomes themselves are very rare, which is confirmed by findings from the analysis of genetic amniocentesis data [37,40].
Considering the mechanisms of X tetrasomy and pentasomy development, a potential explanation of this rarity is possible. In contrast to the common sex chromosome aneuploidies (45,X, 47,XXY, 47,XYY, 47,XXX), which result from a single error in cell division, X tetrasomy and pentasomy involve more than one error, it is clear that the probability of occurrence of multiple errors is much lower.
Thus, the description of each new case of tetrasomy X, an extremely rare pathology, is very interesting and necessary in order to specify the most characteristic features of the phenotype peculiarities.
Considering that literature data regarding the presence of POI in patients with tetrasomy X is even more scant and tetrasomy X has not been detected by cytogenetic examination among patients with POI [41,42], it is clear that our described case is very interesting. Analysis of literature data shows that a large portion of individuals are diagnosed with tetrasomy of X chromosome, in cases when they are referred to physicians due to psychomotor development defects [4,6,12,16,17].
Finally, the identification of POI in cases of tetrasomy X since early puberty is of great importance for timely replacement therapy with estrogens, which causes breast development, arrests longitudinal growth, and stimulates bone formation and prevents osteoporosis.
Conclusion
For the diagnosis of tetrasomy X in patients with POI and psychomotor retardation, karyotyping is advisable despite the scarcity of minor physical anomalies. Timely initiation of estrogen replacement therapy from early puberty in such patients is important, as it ensures the completion of pubertal development, control of longitudinal growth and the prevention of negative long-term consequences of estrogen deficiency, including osteoporosis.
Declaration of interest statement: The authors report having no conflicts of interest
Informed consent: Informed consent forms have been obtained by authors from the patient’s parents for imaging and other clinical information reported in the present article.