Introduction
Leydig cell tumor is an uncommon clinical entity and accounts for less than 0.1% of all ovarian tumors [1, 2]. These tumors are usually unilateral and benign [3, 4]. Leydig cell tumors are functional tumors, which lead to hyperandrogenism and virilization as a consequence of high production of testosterone. Symptoms and signs may include hirsutism, male pattern alopecia, increased muscle mass, voice hoarseness, and clitoromegaly. These tumors present mostly in postmenopausal women [1, 2, 5].
On the other hand, primary hyperparathyroidism (PHPT) is defined as hypercalcemia with increased or inappropriately normal plasma parathyroid hormone (PTH) levels. It is three to fourfold more frequent in women, and occurs after the age of 50 years [6]. PTHP usually presents as an isolated, sporadic clinical entity, but in approximately 10% of cases, it presents with a clear hereditary pattern and sometimes in association with other hormone-producing tumors or other characteristic clinical features [7]. Such is the case of syndromes like multiple endocrine neoplasia (MEN), hyperparathyroidism-jaw tumor syndrome (HPT-JT), familial hypocalciuric hypercalcemia, and familial isolated hyperparathyroidism.
The literature to date reports only one case of association between rare ovarian Leydig cell tumor and PHPT [8]. Herein, we report the case of a postmenopausal woman presenting with virilization due to an ovarian Leydig cell tumor and PHPT.
Case report
A 59-year-old female with a history of breast cancer, obesity, well controlled type 2 diabetes on metformin, primary hypothyroidism, and hypertension presented to the endocrinology clinic with chief complaints of hirsutism and male pattern hair loss. As regards her reproductive history, she experienced menarche at the age of 14 and reported having had regular menstrual periods during her reproductive age. She claimed no knowledge or recollection of any history of hyperandrogenism or ambiguous genitalia as a newborn or in early childhood, and reported normal development of secondary sexual characteristics during puberty. She used oral contraceptive pills while sexually active when younger. She married in her mid-40s, but was never able to conceive. Symptoms of hyperandrogenism started after the menopause, which she experienced in her early to mid-40s. She was found to have elevated testosterone levels and was started on spironolactone. After 2 years the medication was discontinued due to lack of clinical improvement and the patient had no further follow up until she began care at our center approximately 10 years later. She denied the use of systemic corticosteroids, androgens, or anabolic steroids. Physical examination was relevant for clear evidence of male pattern alopecia and hirsutism. She showed no evidence of supraclavicular or dorsocervical fat or any other cushingoid features.
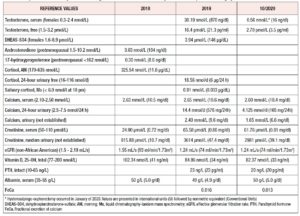
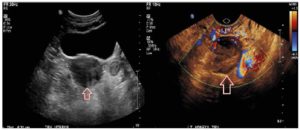
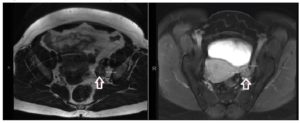
Further workup revealed significantly elevated total testosterone levels, i.e., in the 30s nmol/L (normal range in females: 0.3 to 2.4 nmol/L), with high free testosterone since 2007. Normal dehydroepiandrosterone sulfate, androstenedione, 17-hydroxyprogesterone, 24-hour urine cortisol, late night salivary cortisol, and proper suppression on the 1 mg dexamethasone suppression test (Table 1). After 1 year of repeated imaging studies, she was found to have a hypoechoic lesion in the left ovary on pelvic ultrasound (Figure 1). Abdominal/pelvic MRI also demonstrated a mildly enlarged ovary with hypointense T2 signal (Figure 2). She underwent bilateral hysterosalpingo-oophorectomy and the pathology revealed a 2.2 cm Leydig cell tumor, nonhilar type, in the left ovary. Immunohistochemistry was positive for inhibin, calretinin, and steroidogenic factor-1. In addition, intraoperatively the patient was found to have Leiomyomata uteri, up to 1.6 cm, atrophic endometrium and unremarkable cervix, right ovary, and bilateral Fallopian tubes. The patient reported improvement of the hyperandrogenic symptoms postoperatively and serum testosterone and free testosterone levels returned to normal (0.56 nmol/L and 2.70 pmol/L respectively).
In addition, the patient was noted to have hypercalcemia on different serum measurements, with normal serum albumin, normal glomerular filtration rate, normal vitamin D, and inappropriately normal PTH. 24-hour urine calcium measurement showed significantly elevated calcium in the urine (14.4 mmol/d) with a fractional excretion of calcium of 0.16, consistent with PHPT. She had normal bone density on dual-energy X-ray absorptiometry (DEXA), no history of nephrolithiasis or pathologic fractures, and, to her knowledge, no family history of hyperparathyroidism.
Discussion
Ovarian sex cord-stromal tumors (SCSTs) are a group of benign and malignant neoplasms that develop from the sex cord, stromal cells or both (e.g., Sertoli-Leydig cell tumor) [9, 10]. Ovarian SCSTs are unusual neoplasms that account for approximately 7% of all ovarian tumors [11]. In the 2014 World Health Organization classification scheme for ovarian SCSTs, Leydig cell tumors are included in the group of pure stromal tumors. Leydig cell tumors are very rare neoplasms that account for 0.1% of all ovarian tumors; they present at an average age of 43 years and are commonly benign [12]. Leydig cell tumors occur usually in postmenopausal women, who mostly exhibit hyperandrogenism [13]. Therefore, although other benign androgen excess disorders, such as polycystic ovarian syndrome (PCOS) and idiopathic hirsutism, are frequent, ovarian mass should be part of the differential diagnosis [14].
Most cases of hirsutism are secondary to androgen excess (≥80%), and the majority of women with hirsutism (>70%) have PCOS. Some women (5% to 20%) have “idiopathic hirsutism”, which means hirsutism without hyperandrogenemia. Infrequent causes of androgen overproduction should also be considered, such as non-classical congenital adrenal hyperplasia (NCCAH), which occurs in 4.2% of cases, and androgen-secreting tumors, which are present in 0.2% of hyperandrogenic women; over half of these tumors are malignant. Differential diagnosis of hirsutism should also include Cushing syndrome, acromegaly, hypothyroidism, hyperprolactinemia, and use of topical androgens, exogenous androgens, anabolic steroids or valproic acid [15].
The evaluation of a postmenopausal woman with hyperandrogenism should start with a biochemical workup to rule out other possible etiologies (e.g., Cushing syndrome, adrenal mass, etc.). All women with hirsutism should be tested for elevated androgen levels. Therefore, early morning measurements of serum total and free testosterone should be obtained. Furthermore, early morning 17-hydroxyprogesterone levels should be obtained in order to screen women for NCCAH due to 21-hydroxylase deficiency; this can be done by measuring levels on a random day for those with amenorrhea [15].
In the right clinical scenario, when hyperandrogenism of ovarian source is suspected, imaging studies should be ordered to help identify the tumor after completion of initial hormonal workup. Transvaginal ultrasound (US) with color Doppler and MRI can help identify these tumors, which are usually characterized by morphologic changes within the ovary [16]. Leydig cell tumors appear as unilateral solid masses during imaging evaluations. They tend to be small (mean 2.4 cm) and are isoechoic on transvaginal US and hypoattenuating on computed tomography (CT) [12, 17, 18]. The signal intensity on T2-weighted imaging differs, depending on the amount of fibrous stroma [18, 19]. In T1- and T2-weighted images areas of hyperintensity may be noticed, which reflect the presence of lipid components [19]. Sometimes, if they are particularly small, these tumors are difficult to identify on CT scans and transabdominal US, and repeated imaging or even invasive diagnostic procedures like ovarian and adrenal vein sampling may be necessary. Venous sampling has a key role in the diagnosis of androgen-producing tumors in situations where peripheral testosterone concentration is elevated and imaging studies are not revealing. This procedure can correctly localize 66% of these tumors [20]. The small size of the tumor probably explains why it was not detected on initial imaging evaluation in our patient, despite her displaying clear evidence of virilization and elevated levels of testosterone.
Unfortunately, clinical presentation and sonographic appearance cannot predict whether a mass is malignant [21]. Therefore, surgery is the best next step in the assessment of an ovarian mass. Surgery is the main treatment of ovarian SCSTs, and is also diagnostic [3]. Comprehensive staging occurs intraoperatively and involves examination of the contralateral ovary, palpation of lymph nodes (with resection if there are concerning features), sampling of peritoneal fluid, omental biopsy, and biopsies of the peritoneum and any suspicious lesions. The procedure excludes complete lymphadenectomy as the risk of nodal metastasis with primary disease is low [22].
Pathology reports show that Leydig cells stain for calretinin, inhibin, and melan-A, and they are commonly negative for epithelial membrane antigen and keratin [23]. Leydig cell tumor (previously subclassified as Pure Leydig cell tumor - nonhilar type as per the former WHO classification 2003) is derived from ovarian stroma, which show stromal hyperplasia and contains foci of lutein-like stromal cells [24].
PHPT is the most likely diagnosis when hypercalcemia and elevated or inappropriately normal parathyroid hormone levels are present [25]. The differential diagnosis of hypercalcemia includes conditions which mimic PHPT, including familial hypocalciuric hypercalcemia (FHH), tertiary hyperparathyroidism, and medication use, such as use of lithium or hydrochlorothiazide [25]. Twenty-four-hour urine calcium measurement will assist in the differential diagnosis of FHH. Advances in genetics have led to an estimate that 10% of patients with PHPT harbor a mutation in one of 11 genes that have been associated with syndromic and non-syndromic forms of PHPT [26]. In tertiary hyperparathyroidism, parathyroid glands assume a quasi-autonomous role, similar to what occurs in PHPT. However, differentiation of tertiary hyperparathyroidism from PHPT is based on a clearly identifiable history of longstanding secondary hyperparathyroidism as a consequence of malabsorption or renal failure preceding the onset of hypercalcemia [27].
Hypercalciuria occurred in approximately 40% of 70 patients with PHPT in a longitudinal study [28]. Hypercalciuria helps in identifying PHPT from familial hypocalciuric hypercalcemia, which presents, similarly, with hypercalcemia and minimally elevated PTH or levels in the mid-upper normal range [29]. PHPT is identified earlier in countries with routine screening and may be asymptomatic. On the contrary, where routine biochemical testing is not available, PHPT is more likely to present with nephrolithiasis and/or skeletal complications [6].
Parathyroidectomy is indicated, and is the preferred treatment, for all patients with symptomatic PHPT, those with serum calcium levels greater than 0.25 mmol/L (1.0 mg/dL) above normal, regardless of symptoms, and when there is objective evidence of renal involvement (e.g., nephrolithiasis, nephrocalcinosis, hypercalciuria, or glomerular filtration rate <60 ml/min), osteoporosis, fragility fracture, evidence of vertebral compression fracture, or evidence consistent with parathyroid cancer [30].
In the present case, our patient did not meet surgical criteria for her diagnosis of PHPT, and is being managed conservatively with close monitoring. She presents with a normal DEXA scan, despite having longstanding hypercalcemia. This phenomenon could be explained by the protective effects of high levels of testosterone with subsequent bone aromatization to estradiol, thus decreasing bone resorption. Severe osteoporosis has been noticed in several case reports involving men with mutations in the estrogen receptor or aromatase genes [31, 32], indicating that the effects of testosterone on the skeleton, at least in part, are mediated by its aromatization to estradiol [33].
Ovarian tumors, such as ovarian epithelial carcinomas, occasionally present with severe hypercalcemia. Common causes include PTH-mediated hypercalcemia and PTH-related peptide (PTHrP)-induced humoral hypercalcemia of malignancy (PTHrP-HHM). Although ectopic hyperparathyroidism caused by tumor cells is extremely rare, PHPT due to parathyroid adenoma or hyperplasia occurs more frequently [34]. In PTHrP-HHM, severe hypercalcemia is evident and PTH is suppressed.
Parathyroid imaging is not a diagnostic procedure for PHPT and is not advised unless surgical intervention is planned [6]. Thus, it has become a standard preoperative procedure to localize abnormal parathyroid tissue [25]. The only curative approach is parathyroidectomy when indicated. After the diagnosis of PHPT has been made, familial disorders should be considered, such as MEN types 1, 2A, and 4, HPT-JT, and familial isolated hyperparathyroidism [35]. Thus far, we have been able to identify a limited number of the genes involved in these syndromes. CASR, MEN1, CDNK1B, and CDC73/HRPT2 are probably the most common ones. However, in a significant number of cases, no identifiable mutation is found, suggesting that new genes involved are yet to be discovered. Even though our patient had uterine fibroids (leiomyomata), her condition is unlikely to be HPT-JT, as this condition appears to have a more aggressive course, with patients tending to have more severe hypercalcemia [35]. This was not the case of our patient, who showed mild hypercalcemia.
To date, there is only one previous report of association between an ovarian Leydig cell tumor and PHPT due to a parathyroid adenoma in a postmenopausal female patient [8]. Unlike our case, this latter patient’s PHPT was present in the context of higher levels of hypercalcemia and elevated serum PTH. The concurrence of a virilizing ovarian tumor and PHPT may be an exceptional association, or it could be part of a still undefined syndrome due to an inherited or sporadic mutation.
Syndrome such as MEN have been largely described in literature. MEN is described by the occurrence of tumors involving two or more endocrine glands within a single individual. MEN may be inherited as autosomal-dominant syndromes or may occur sporadically. Furthermore, MEN4, also referred to as MENX, occurs due to cyclin-dependent kinase inhibitor (CDNK1B) mutations. MEN4 is characterized by the presence of parathyroid and anterior pituitary tumors in association with tumors of the kidneys, adrenals, and reproductive organs [36].
Conclusion
We are presenting this case because of the rare association of ovarian Leydig cell tumor and PHPT in a postmenopausal female patient with virilizing symptoms, which appears to be the second such case reported in the literature to date. This could be a rare coincidence or linked to an inherited or sporadic mutation yet to be discovered. This case underlines the concept that in the presence of hirsutism, rare etiologies should be sought. Virilizing tumors of the ovary should be considered despite negative initial imaging studies, which should eventually be repeated if virilizing symptoms persist. PTHP should be considered in patients with asymptomatic hypercalcemia (e.g., normal DEXA scan) with inappropriate normal PTH and hypercalciuria.
Acknowledgment
No funding.
Conflict of Interest Statement:
The authors declare that there is no conflict of interest.