Introduction
Premature ovarian insufficiency (POI) is a disease characterized by oligomenorrhea, hypoestrogenism and a follicle-stimulating hormone (FSH) level higher than 25 IU/L that occurs in women younger than 40 years of age.[1,2] It has been suggested that POI may affect 1% of women under 40.[1,3,4]
The term POI has replaced the older “premature ovarian failure” because the previous definition alluded to an unexpected and acute occurrence rather than a progressive process.[1,2] Nevertheless, both definitions can still be found in the literature.
There are several causes of POI, including chromosomal and genetic diseases, and autoimmune, infectious or iatrogenic events. Unfortunately, in about 75% of POI cases the cause remains unknown.[1,3,4] Depending on the cause, POI can be classified as primary (spontaneous) or secondary (iatrogenic).
From a pathogenetic point of view, primary ovarian insufficiency occurs through two major mechanisms: follicle dysfunction and follicle depletion.[5] From a clinical perspective, primary POI is a disorder in which a young woman presents menstrual irregularity or amenorrhea.
Although the disease is not particularly frequent in the general population, it is crucial to make an early diagnosis, as the consequences of primary POI, when untreated, differ from those of other disorders characterized by menstrual irregularity, such as polycystic ovarian syndrome or hypothalamic amenorrhea. In fact, POI may lead to osteoporosis, ischemic heart disease, stroke and infertility, as well as depression and anxiety.[6-10]
The aims of this review are to examine the factors that can determine or contribute to primary POI, and to discuss the developments that are likely to be significant in the future, including the desirable avenues of future research.
Methods
In this narrative review, we provide an overview of possible factors associated with primary POI.
We carried out a search in the PubMed database to identify eligible studies published up to April 30th, 2020. The search was performed using the following terms: “primary ovarian insufficiency”, “POI”, “premature ovarian insufficiency”, “POF”, “premature ovarian failure”, “premature hypergonadotropic hypogonadism”.
We analyzed randomized clinical trials in humans, together with retrospective, comparative and observational studies, meta-analyses and reviews, whereas we excluded case reports and papers that were not written in English. We then evaluated the references cited in all the papers considered with the aim of identifying possible additional eligible articles. The final number of studies included in our review was 93.
Results
Genetic causes
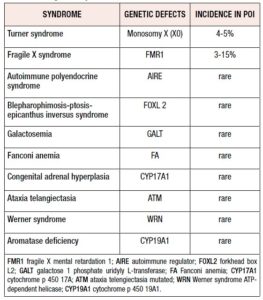
POI can be associated with syndromic diseases as illustrated in Table I.
Genes localized in the X chromosome play a crucial role in ovarian development and functioning, as demonstrated by studies regarding women affected by Turner syndrome; however some autosomal genes can also be involved in the pathogenesis of POI.[11-16] Furthermore, even rare, isolated POI without syndromic manifestations has been associated with single-gene mutations (such as in DIAPH2, MBP15, BRCA2 and STAG3).[17-21] There is some evidence for a suspected role of genetic alterations in cases of idiopathic POI.
Turner syndrome
Turner syndrome is caused by the most common chromosomal abnormality in females. It is clinically characterized by skeletal abnormalities such as short stature, micrognathia, cubitus valgus, short neck, scoliosis, lymphedema of hands and feet, ovarian failure and cardiovascular diseases.[11]
The incidence of Turner syndrome is 1:2,500 live births in Western countries, and the disease accounts for 4-5% of POI cases.[11,12,22]
The chromosomal defect in Turner’s syndrome is partial or total absence of the X chromosome. In 45% of cases, it is characterized by the genotype 45X0, and in 50% by mosaicism for 45X, but several types of X anomalies can also be involved (e.g., isochromosome Xq, deletion of Xp or Xq).[22,23]
The short stature in Turner’s syndrome is caused by haploinsufficiency of the SHOX (short stature homeobox) gene, located on both the X and the Y chromosomes, which has also been linked to short stature in Leri-Weill dyschondrosteosis and idiopathic short stature.[24,25]
Moreover, the X chromosome contains critical genes involved in ovarian functions, such as BMP15 (“bone morphogenetic protein 15”) on the short arm of the X chromosome, and FMR1 and FMR2 (“fragile X mental retardation protein”) on the long arm, [26] and aneuploidy may cause a reduction in the number and survival of oocytes.[23]
In a 1966 study, [27] histological sections of the gonads of thirteen 45,X0 abortuses were compared with sections of gonads from diploid XX specimens and no difference was found up to the third intrauterine month, while in the older X0 fetuses an increase of connective tissue was found as the demonstration of accelerated follicle depletion.
A wide range of gonadal dysfunctions is observed in Turner syndrome. Most affected women suffer from primary amenorrhea and absence of thelarche, while others can initially develop regular menses.
In an Italian retrospective multicenter study in 522 women with Turner syndrome, 16.1% had spontaneous pubertal development and menarche; 30 patients had regular menses five years after their menarche, 12 had secondary amenorrhea, and 19 had irregular menstrual cycles. Pregnancy occurred in 3 patients (3.6%).[28]
The 2015 European Society of Endocrinology Guidelines recommend growth hormone (GH) treatment in 4–6-year-old girls affected by Turner syndrome and showing growth failure or short stature, or oxandrolone from the age of 10 years if the diagnosis of the disease is delayed.[29]
Most affected women need a low-dose transdermal estradiol to induce puberty, to maintain feminization during adult life, and to reduce morbidity and mortality.[30]
This treatment should be started at between 11 and 12 years of age if there is no evidence of breast development, and progesterone should be added after 2 years of treatment or in the case of breakthrough bleeding to prevent endometrial hyperplasia and the risk of endometrial cancer.[29,31]
Furthermore, the 2015 European Society of Endocrinology guidelines recommend counseling in these young women about the possibility of achieving spontaneous pregnancy or undergoing fertility treatment. Ultrasound imaging and a magnetic resonance scan of the thoracic aorta and heart before planned pregnancy must be obtained, considering the increased cardiovascular risk during pregnancy.[29,32]
In a retrospective case series of 276 women with Turner’s syndrome, 1.4% experienced spontaneous pregnancy, 1.4% achieved pregnancy using assisted reproduction technology, and 0.4% both spontaneous and assisted pregnancy.[33]
Fragile X syndrome
Fragile X syndrome (FXS) is an X-linked disorder characterized by deficiency or absent levels of fragile X mental retardation protein (FMRP), caused by a loss-of-function mutation of the FMR1 gene, located at Xq27.3 and due to an expansion of a trinucleotide (cytosine-guanine-guanine, CGG) repeat in the promoter region. FMRP is an RNA-binding protein implicated in the regulation of mRNAs involved in synaptic connections.[34-36]
A full mutation, defined as an expansion of >200 CGG repeats, leads to the classical phenotype with hypermethylation of FMR1 with transcription silencing and FMRP reduction.
An expansion of between 55 and 200 repeats is known as a ‘premutation’, and it may result in a full mutation in subsequent generations; in this case, in contrast with the full mutation, the FMR1 gene is transcriptionally active and the classic syndrome phenotype does not occur.[36,37]
Fragile X syndrome is clinically characterized by physical, cognitive and behavioral features, such as a long and narrow face, large ears, attention deficit hyperactivity disorder, anxiety, and autism spectrum disorder.[38,39]
The manifestations of the syndrome are variable, depending on age, molecular variation (full mutation or premutation), environmental influences and sex; females have less severe manifestations than males, because of the percentage of cells with the normal FMR1 allele on the active X chromosome that produces FMRP. [36-40]
Individuals with fragile X premutations (55–200 CGG repeats) can develop X-associated tremor-ataxia syndrome, a disorder characterized by intention tremor, cerebellar ataxia and cognitive decline; moreover, according to some authors, 20% of affected women will develop POI.[41,42]
The authors of another study showed that there is a strong relationship between age at menopause, including POI, and premutations of the FMR1 gene: 12 to 28% of women with premutations go through menopause before the age of 40.[43]
Bodega et al.[44] investigated the number of CGG repeat tracts in 190 patients with POI and in a control group of 200 women with physiological menopause, and found a high prevalence of FMR1 premutation (range 63–163 repeats) (19 out of 190, 10%, 95% confidence interval (CI) 5.8–14.2%) in patients with POI compared with the control population (0/200).
Sullivan et al. found significant associations between the number of CGG repeats in the premutation range and ovarian dysfunction and age at menopause.[45]
The exact mechanism by which a premutation causes ovarian failure is not known.
Subjects with premutations appear to have increased FMR1 mRNA expression, which probably leads to toxicity, since they have a normal amount of protein.[36,37,46,47]
By comparison, full mutations are associated with methylation of the FMR1 gene and a lack of transcription and protein production; no association was found between POI and full FMR1 mutations.[36]
In a study evaluating 11 fragile X premutation carriers with normal menses, and 22 age-matched controls, the patients gave daily blood samples for measurement of luteinizing hormone (LH), FSH, estradiol, progesterone, inhibin A, and inhibin B levels. Total cycle and follicular phase length were decreased in fragile X premutation carriers, who showed elevated FSH during the follicular and luteal phases, and decreased inhibin A and progesterone in the luteal phase, whereas there was no difference in estradiol or LH. These hormonal changes suggest that fragile X premutation carriers, despite regular menstrual cycles, exhibit early ovarian aging likely due to decreased follicle number and functioning, as reflected by lower inhibin B, inhibin A, and P4 levels.[48]
On this basis, the American College of Obstetricians and Gynecologists now recommends that women with POI or an elevated FSH level before the age of 40 without known cause be screened for FMR1 premutations.[49]
The 2016 European Society of Human Reproduction and Embryology Guideline recommends fragile-X testing in all women with POI, together with careful counseling, not only to investigate the cause of POI, but also because mutation of the FMR1 gene could have family implications.[50]
Galactosemia
Galactosemia is an autosomal-recessive disorder, whose classic form is caused by deficiency of galactose-1-phosphate uridyl transferase (GALT). It is characterized by liver, kidney, heart and ovarian involvement.[13] The reported incidence in Western countries is 1 in 60,000 live births.[14]
Calderon et al. described more than 200 mutations in the GALT gene.[15]
POI is present in all women with homozygotic mutation of this gene,[13] probably due to the toxic effects of galactose metabolites that cause apoptosis of oocytes.[16]
Blepharophimosis-ptosis-epicanthus inversus syndrome
Blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) is a rare condition caused by mutation of the forkhead box L2 gene (FOXL2) on chromosome 3. BPES is characterized by autosomal dominant eyelid malformation, which may or may not be associated with POF.[51,52]
Other genes associated with POI
A heterozygous mutation in the bone morphogenic protein-15 gene (BMP15) was found in two sisters with POI. BMP15 is a protein involved in folliculogenesis in animal models.[17]
Caburet et al.[18] found a homozygous deletion in the gene encoding stromal antigen 3 (STAG 3), in a consanguineous Palestinian family with hereditary POI. STAG 3 encodes a protein subunit of cohesin, a protein complex that regulates pairing and dissociation of sister chromatid during meiosis.[19]
Furthermore, histological analysis of ovaries in female mice with a homozygous mutation in STAG 3 showed a loss of oocytes indicating early ovarian dysgenesis.[18]
Bione et al. identified a mutation of a human homolog of the Drosophila melanogaster diaphanous gene (DIAPH2) in a family with POI.[20]
A further study [21] reported two sisters with XX gonadal dysgenesis caused by mutations in the BRCA2 at chromosome 13, leading to low protein levels and loss of repair in double-stranded DNA breaks that occurred during meiosis. Such findings indicate that DNA repair genes have a critical role in ovarian development.[53]
Lang-Muritano et al. described a 16-year-old female with osteoporosis, streak ovaries and primary amenorrhea due to a loss-of-function mutation in the estrogen receptor 2 (ER2) gene, with altered signaling of estradiol. This case suggests the importance of ER2 in ovarian function.[54]
Autoimmune ovarian insufficiency
Some authors noted that ovarian insufficiency is correlated with adrenal insufficiency. Indeed, it was observed that some women affected by adrenal insufficiency also had ovarian insufficiency.
This clinical picture of multiple failure includes two different syndromes of polyglandular autoimmune failure: type I and type II. These syndromes are associated with autoantibodies directed against multiple endocrine organs.
Moreover, myasthenia gravis may be associated with POI. In 2004, Ryan et al., reported the case of a woman with seropositive myasthenia gravis who subsequently developed autoimmune POI with antibodies against the receptor for FSH.[55]
In 2010, Li et al. described a patient with myasthenia gravis, whose myasthenic symptoms were precipitated by an estrogen treatment. They supposed that there was likely a common autoimmune disease mechanism, although the precise pathogenesis of these disorders remains partially obscure.[56]
Hoek A et al.,[57] in 1997, reported lymphocytic infiltration of thecal cells in women with POI associated with adrenal autoimmunity and/or Addison's disease. In their study, they showed that these women had autoantibodies against steroid-producing cells.
Furthermore, they also showed an autoimmune pathogenesis in women with idiopathic POI, in the absence of adrenal autoimmunity or Addison's disease. Some immune abnormalities could be the cause of POI in women with IDDM or Graves’ disease.
Smoking
Cigarette smoke contains polycyclic aromatic hydrocarbons (PAHs), which bind to the aryl hydrocarbon receptor present on the surface of granulosa cells. This binding activates the Bax gene and cytochrome P450, which converts PAHs into other toxic substances.[58-60]
These molecules have a toxic effect on the ovary and on reproductive function.
Studies regarding the association between smoking and age at menopause and POI are conflicting: while there is increasing evidence that smoking, being involved in diminished ovarian reserve, influences age at menopause, its impact on the prevalence of POI has not been clearly demonstrated.
Sun et al.[61] showed that cigarette smoking is an independent variable for earlier menopause.
Harlow and Signorello,[62] in 2000, and Parente et al., [63] in 2008, all highlighted an association between cigarette smoking and age at menopause onset in women.
Fleming et al., showed that menopause occurred 0.8–1.4 years earlier in 7596 women who smoked.[64]
In 2015, Tawfik et al. highlighted that cigarette smoking has an odds ratio of 1.8 (1.03–3.23) for the development of POI in women.[65]
Finally, a series of studies conducted by Caserta et al., Chang et al., and Freour et al. showed that cigarette smoking caused a reduction in the number of antral follicles, [66] an increase of FSH levels in blood, [66,67] and a reduction in AMH levels.[68] The number of pack-years also correlated with these data.[66]
Viruses
There is still little evidence that a viral infection may cause POI; indeed, a real cause-and-effect relationship has not yet been established.
In 1975, Morrison et al. described three women with premature menopause presumably caused by mumps oophoritis.[69]
Subietas et al. described three postmenopausal patients who had an ovarian infection at autopsy caused by cytomegalovirus (CMV). They did not highlight clinical evidence of hypogonadism.[70]
Role of vaccination
There has been concern in the past about a possible correlation between primary POI and human papillomavirus (HPV) vaccination, albeit deriving only from isolated case reports.
A 2012 case report described a 16-year-old woman with alteration of menses followed by a diagnosis of POI, after papillomavirus vaccination with a quadrivalent HPV vaccine.[71]
Furthermore, Little and Ward reported three teenagers with POI who had received quadrivalent HPV vaccination. [72]
Similarly, Colafrancesco et al. documented the history of three women who developed POI following HPV vaccination.[73]
However, there is one retrospective cohort study which investigates the association between POI and vaccines. This study included 199,078 women who had received at least one dose of HPV vaccine. No statistically significant increased risk of POI (HR 0.30, 95% CI: 0.07–1.36) was found after administration of HPV, Tdap (tetanus, diphtheria, pertussis), influenza, or meningococcus vaccination.[74]
Environmental pollutants
At present, there is considerable interest in the possible influence on ovarian function of three different factors: endocrine-disrupting chemicals (EDCs), oxidative stress and epigenetics.
Endocrine-disrupting chemicals are exogenous chemical molecules, or mixtures of chemicals, that interfere with hormone function, at any level. Their effects may be permanent or transitory.
Oxidative stress means elevated reactive oxygen species (ROS) production. When elevated ROS production occurs in ovarian cells, it can damage ovarian function, determining, together with other factors, the initiation of antral follicle apoptosis.[75]
In the presence of epigenetic modifications, DNA methylation alters ovarian function. These modifications may be inherited by subsequent generations.[76-78]
With regard to EDCs, more than 20 substances involved in the development of POI have been identified: four pesticides, three solvents, five compounds used in industrial chemistry, three metals (cadmium, lead, chromium), three compounds belonging to the family of polycyclic aromatic hydrocarbons, methoxychlor (MXC) and bisphenol A.[79]
A further 15 substances were also suggested to be related to the development of POI, but the evidence was insufficient. These substances may cause reduction of the follicle pool.[79]
Phthalates, substances that remain in the environment for several years,[80] are endocrine disruptors that influence ovarian functioning. The ovarian toxicity of phthalates on the ovary is linked to their effects on folliculogenesis and steroidogenesis.[81]
Bisphenol A (BPA) is an estrogen mimicker that is able to interact with estrogen receptor alpha and alter endocrine function.[58] In 2014, Richardson et al.,[58] Machtinger and Orvieto, [82] and Caserta et al.[66] showed that BPA was associated with alteration of ovarian reserve.
Exposure to herbicides and pesticides, for example simazine or MXC, can determine a decrease in total ovary weight, an element of undefined significance.[83,84]
An epidemiological study comprising 8038 women, conducted by Farr et al. in 2006, showed that the median age at menopause was increased by 3 to 5 months in women who lived and worked on American farms, depending on the types of pesticides used.[85]
A case-control study of 1407 women, conducted by Cooper et al., highlighted that a higher concentration in plasma of 1,1- dichloro-2,2-bis(4-chlorophenyl) ethene, a dichlorodi-phenyldichloroethylene isomer, could lower the age at menopause.[86]
Pan et al. showed that exposure to polychlorinated biphenyls (PCBs) and dichlorodiphenyltrichloroethane (DDT) could be a potential risk factor for POI, with women exposed to PCBs showing higher levels of LH, and those exposed to DDT lower levels of AMH.[87]
Zhang et al., in 2018, highlighted that the exposure to perfluorooctanate, perfluorooctane sulfonate, and perfluorohexanesulfonate in 120 Chinese women was associated with increased risk of POI.[88]
Primary hypogonadism without follicle depletion
There are other conditions in which genetic alterations of estradiol precursor production or aromatase function may cause POI.
These disorders reduce the production of estradiol and determine a lack of correct negative feedback on the FSH hormone. Such disorders cause primary hypogonadism. They are not associated with follicular depletion but are related to an increase of circulating FSH.
Intraovarian modulators
Some paracrine substances can regulate ovarian reactivity to gonadotropins. This regulation can be direct or indirect, resulting in reduced responsiveness of the ovary to gonadotropins. At present, only polymorphisms in the alpha subunit of inhibin have been identified as a possible cause of POI.
The possible role of inhibin alpha gene variants in POI was studied by Shelling et al. in 2001. They studied 43 women with POI for mutations in the three inhibin genes. They found two variants: a 1032C-->T transition in the INHssA gene in one woman, and a 769G-->A transition in the INHalpha gene in three women.[89]
Steroidogenic enzyme defects
Some uncommon cases of POI may be related to genetic defects of the enzymes involved in the biosynthesis of androstenedione, estradiol and cortisol. These disorders cause low circulating estrogen levels and, consequently, high serum FSH levels. As a consequence, notwithstanding their normal follicle pool, these patients present hypergonadrotropic hypogonadism.
The genes coding for these enzymes, and found to show mutations, are steroidogenic acute regulatory enzyme (StAR),[90,91] CYP17, aromatase gene, [92] and NR5A1 (steroidogenic factor 1).[93,94]
FSH receptor mutations
Some cases of POI have been associated with defects in FSH receptor (FSHR) production. These alterations lead to ineffective binding of circulating FSH with its receptor. Consequently, in some of these cases, FSH did not activate the aromatization of estradiol precursors. Hence, these patients were deficient in estrogens.
This condition was studied by Aittomäki et al. in 1994 and 1995, and by Nakamura et al. in 2008.
Aittomäki mapped a locus for ovarian dysgenesis to chromosome 2p and found the mutation in a C566T transition in exon 7 of FSHR.[95]
Nakamura et al. mapped a new heterozygous 662T->G mutation in exon 8 in a 25-year-old woman with primary amenorrhea.[96]
The possible consequences on the ovarian pool of estrogen deficiency and the subsequent effects on fertility are far from clear.
Gs alpha subunit gene mutations
Other gene mutations possibly linked indirectly to POI may be guanine mutations of adenylate cyclase (protein G), which may result in alterations of adenylate cyclase activity. Patients carrying these mutations may have an abnormal gonadal function, as protein G usually has a role in activating FSH and LH receptors.[97-99]
Conclusion
In this narrative review, we have provided an overview of possible causes associated with primary POI. There are many possible causes of POI, including chromosomal and genetic diseases, and autoimmune, infectious or iatrogenic events. In about 75% of POI cases, however, the cause remains unknown. Untreated POI may lead to estrogen deficiency complications such as osteoporosis, ischemic heart disease, stroke and infertility. Early diagnosis of POI is a challenge for gynecologists, but recognizing the cause is the main issue. Indeed, in selected cases, diagnosis of the possible cause leading to POI has important implications regarding the treatment of related comorbidities, family screening, and appropriate counseling, also with the aim of prevention.
This literature overview underlines that early diagnosis is key in the management of young patients suffering from primary POI, as it allows the establishment of a proper hormone replacement therapy.
Among our group of POI patients referred to the Infertility Center of Pisa University Hospital from January 2013 to December 2020, 51 women were evaluated. The median age of these women was 37.5 years. Genetic counseling did not identify a specific genetic cause of POI in these cases and all were found to have a normal karyotype. Therefore, no further in-depth analyses were recommended. Among the described factors that may hypothetically lead to POI, HPV vaccination had been performed in 4/51 (7.8%) of the patients, whereas previous CMV infection was detected in 5/51 (9.8%). Notably, smoking (≥ 5 cigarettes/day for ≥ one year, currently or in the past) was reported in 15/51 (29.4%) of our POI patients.
At present, there is a lack of availability of reliable and affordable genetic and molecular tests in public infertility centers, and the investigation of environmental pollutant agents is still challenging.
As a consequence, the only directly modifiable factor leading to POI in the general population is cigarette smoking, making it crucial to target our efforts against this habit.
Patients with primary POI must be informed and counseled with regard to their fertility preservation options. In the future, genetic therapies may allow an individualized approach and tailoring of therapies.
Declaration of interest statement
The authors declare that there is no conflict of interest.