Introduction
Polycystic ovary syndrome (PCOS) is a complex endocrine disorder characterized by oligo-anovulation, hyperandrogenism, and polycystic ovaries on ultrasound. Frequently, PCOS is accompanied by carbohydrate and lipid metabolism disturbances and inflammatory abnormalities that lead to an increased risk of development of cardiovascular disease (CVD) [1-3]. Higher levels of circulating androgens occur in 70-80% of women with PCOS; overweight/obesity and insulin resistance are also found in about 70% of women with this condition [4-7]. The amount and distribution of adipose tissue and the preferential visceral adipocyte deposition seen in normal-weight and overweight/obese women with PCOS are currently not completely understood [8]. Despite the understanding that high levels of androgens help determine visceral adipocyte accumulation in women with PCOS, many knowledge gaps need to be filled [9].
Material and methods
This comprehensive review aimed to summarize the current knowledge regarding the implications of the intimate relationship between androgens and adipocytes, favoring the development of comorbidities in women with PCOS. The review entailed a detailed analysis of publications on the interrelationship between hyperandrogenism and adipocyte dysfunction in women with PCOS. We searched Scopus, Google Scholar, Medline, and PubMed to identify the most relevant publications between 2000 and 2021. The search was expanded by the retrieval of bibliographic citations from the identified articles.
When providing crucial basic knowledge, studies published before 2000 were also included. Keywords used in the search were as follows: polycystic ovary syndrome, hyperandrogenism, obesity, adiposity, dysfunctional adipocyte, insulin resistance, adipose tissue distribution, and various combinations of these terms.
Adipocyte steroid uptake, production,
and metabolism
Adipocytes, directly and indirectly, participate in androgen production [10]. In addition to ovarian and adrenal sources, conversion of the weak androgen dehydroepiandrosterone (DHEA) into androstenedione (A4), of A4 into testosterone (T), and of T into dihydrotestosterone (DHT) in adipose tissue are also essential sources of circulating androgens. Adipocytes express several steroidogenic enzymes, and they locally modulate steroid levels [11]. The steroidogenic enzyme activity of adipocytes appears to be relevant in the development of hyperandrogenism in PCOS, particularly because weak androgens such as A4 are rapidly taken up, sequestered from circulation by mature adipocytes, and converted into strong androgens [10-12]. For this reason, the amount of androgens in adipocytes, which differs in omental and subcutaneous depots, is even more significant than the amount in circulation [13]. Furthermore, enhanced adipocyte steroidogenesis in women with PCOS may explain the association between hyperandrogenism and obesity [14, 15].
Regarding the direct participation of adipocytes in steroidogenesis, levels of type 5 17β-hydroxysteroid dehydrogenase (17β-HSD), the enzyme that converts A4 into T, are increased in subcutaneous adipose tissue of obese women [16]. The activity of 5α-reductase, the enzyme that converts T into DHT, is increased in PCOS with or without obesity [17].
Of note, 17α-hydroxylase (CYP17A1) activity positively correlates with the waist/height ratio and negatively correlates with the conicity index in women with PCOS [18]. Furthermore, 17,20 lyase activity correlates with various biomarkers of adiposity in PCOS [18]. Levels of 11β-hydroxylase are slightly decreased in hyperandrogenemic women with PCOS [19]. These findings suggest that clinical biomarkers of body fat mass and distribution correlate with the activities of various steroidogenic enzymes within adipose tissue.
Indirect participation of adipocytes in the production of androgens may be modulated by the action of adipocytokines on adrenal and ovarian steroidogenic cells. The effects of adipocytokines on steroidogenesis have recently been reviewed [20]. In women with PCOS, there is increased secretion of harmful adipocytokines such as leptin and tumor necrosis factor-α (TNF-α), and lower secretion of beneficial adipocytokines such as adiponectin and omentin-1. The effects of adipocytokines on adrenal cell steroidogenesis depend on their amounts and types. In humans, leptin decreased adrenocorticotropic hormone-induced secretion of DHEA by decreasing the expression of CYP17A1 [21], while adiponectin stimulates cortisol production and the expression of a steroidogenic acute regulatory protein (StAR) and a cholesterol side-chain cleavage enzyme (CYP11A1) [22]. In ovarian cells, there is a bidirectional relationship between theca cells, granulosa cells, and adipocytes [20].
In women with PCOS, leptin levels are positively correlated with T, body fat mass, body mass index, waist-hip ratio, and metabolic abnormalities [23]. Adiponectin has lower levels and is negatively correlated with androgen levels in women with or without PCOS 24]. Resistin levels positively correlate with T through the higher activity of CYP17A1 [25]. In obese PCOS, chemerin and RBP-4 levels are positively associated with T concentrations [26, 27].
Overall, androgens are produced and interconverted by adipocytes, and these phenomena could account for a third source of androgen production, mainly in conditions of adiposity [28]. Regional differences in steroidogenic enzyme activities may exist. The net amount of excessive androgen production, particularly of potent androgens, by adipocytes in women with PCOS has not yet been determined.
The role of androgens on adipocyte function
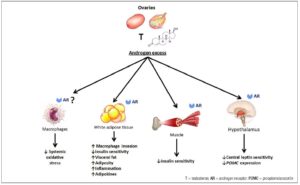
The effect of androgens on adipose tissue function depends on circulating androgen levels, androgen receptor (AR) density and affinity, and local steroidogenic enzyme activities. There are androgen receptors both on preadipocytes and on mature adipocytes [29]. In these cells, androgens may influence gene expression, cell proliferation, cell differentiation, and carbohydrate and lipid metabolism. Androgens increase the transcription of their receptors in mononuclear cells, favoring the release of adipocytokines [30]. Furthermore, T also increases the transcription of the chemokine (C-C motif) ligand 2 (CCL2) in omental adipose tissue, triggering macrophage invasion [31].
As regards gene expression, androgens bound to AR function as transcription factors that regulate the expression of various genes. Gene expression regulation is controlled through several factors [32] that may modulate the transcription of peroxisome proliferator-activated receptor-gamma (PPARγ) and the mitogen-activated protein kinase (MAPK) cascade involved in adipocyte differentiation, proliferation, adipogenesis, and lipid metabolism [33,34]. T also promotes visceral adipose tissue (VAT) distribution and chronic low-grade inflammation (Fig. 1).
The androgen DHT may not inhibit the proliferation of preadipocytes in vitro [35]; however, in general, androgens suppress adipocyte differentiation into mature adipocytes by epigenetic control through decreased PPARγ transcription [36]. Thus, T and DHT inhibit preadipocyte differentiation and late-stage adipocyte maturation in both subcutaneous adipose tissue (SAT) and VAT compartments [37]. By contrast, DHEA has a more complex role [37]. It does not affect subcutaneous preadipocyte differentiation; instead, DHEA decreases adipogenesis in omental preadipocytes [37, 38]. In addition to modulating adipocyte proliferation and differentiation, T at higher levels promotes adipocyte hypertrophy [39] and overproduction of adipokines [40] through AR stimulation [30]. Furthermore, T enhances the infiltration of macrophages of dipocytes and promotes expansion and lipid accumulation in VAT [41]. The distribution and capacity of androgen receptors on adipocytes vary by region, with a higher binding capacity observed in VAT. As regards lipid metabolism, T is a potent regulator of lipolysis and its action varies across species [42]. T modulates adipogenesis and lipolysis. Androgens may increase abdominal lipolysis without altering lipolysis in the gluteal-femoral compartment, by decreasing lipid uptake and synthesis [43, 44]. T diminishes lipolysis in SAT by decreasing expression of β2-adrenergic receptors and hormone-sensitive lipase (HSL), while cyclic adenosine monophosphate stimulates adenylate cyclase activities [35]. At high levels, T diminishes lipolysis in VAT of nonhuman primates [45] and stimulates lipogenesis in women with or without PCOS [2, 46]. Nevertheless, even in nonobese PCOS, T increases the ability of catecholamines to activate hydrolysis of triglycerides into free fatty acids (FFAs) [47]. This primary lipolytic defect in PCOS is an exception.
Adipocyte size in PCOS
The size of adipocytes is closely associated with their function;
hypertrophic adipocytes are dysfunctional [48]. In hyperandrogenic
obese women with PCOS, adipocytes are even larger
than those taken from obese controls [39], and they produce many
adipocytokines [49]. In nonobese hyperandrogenic women with
PCOS, adipocytes may also be enlarged and dysfunctional [50].
The hypertrophy of adipocytes in PCOS may be due to imbalanced
abilities of adipocyte storage or adipocyte lipolytic activities
[51]. Adipocyte size is related to insulin growth factor-1,
phosphoinositide 3-kinase, and protein kinase B gene expression
[52]. Of note, these genes combine external signals such as
insulin and T [52, 53].
In PCOS, hyperandrogenism has an essential role in the development
of adipocyte hypertrophy in SAT and fat expansion
[40, 51, 54]. In obesity, adipocytes are hypertrophic, possessing a
propensity to pro-inflammatory gene expression. They are also
dysregulated in their expression of cytokines and are characterized
by an increased proportion of macrophage invasion [10,55].
Hypertrophic adiposity driven by T is highly correlated with
insulin resistance [39,50]. Hypertrophic adipocytes produce and
secrete various paracrine growth factors that stimulate increases
in the number and size of preadipocytes [56]. In general, because
of the higher production of adipocytokines and cytokines, hypertrophic
adipocytes are associated with abnormal lipid metabolism,
metabolic syndrome, type 2 diabetes mellitus (T2DM),
non-alcoholic fatty liver disease, non-alcoholic steatohepatitis,
and CVD [39,57]. In summary, hypertrophic adipocytes are associated
with PCOS and various comorbid conditions [25, 50, 51].
Adipose tissue distribution in PCOS
In normally-cycling women, various genetic, ethnic, social,
and hormone factors influence body adipose tissue distribution.
Typically, women with a higher percentage of body fat tend to
show distribution of adipose tissue in the hips and thighs [58]. In
abnormal conditions such as obesity and PCOS, the capacity of
subcutaneous adipose tissue to safely store fat is exceeded, and
excess fat is deposited in abnormal locations such as the retroperitoneum, omentum, liver, and muscle [48, 59]. Also, impaired
adipocyte proliferation or differentiation capacity leads to the
redistribution of fat tissue from subcutaneous areas to visceral
depots [51]. In this manner, androgens may drive adipose tissue
mass in a depot-specific way through site-specific modulation
of preadipocyte proliferation and differentiation or by lipid synthesis
(lipogenesis) or lipolysis in mature adipocytes [60].
In women with PCOS, hyperandrogenism modifies body fat
distribution, causing adipocytes to accumulate in the abdominal
wall, and to surround or infiltrate intra-abdominal organs
[46, 61]. Androgens are associated with increased fat mass and
an increased amount of fat localized on the trunk [62, 63]. Thus,
women with PCOS have more central obesity than weight- and
body mass index-matched controls [64]. Total intra-abdominal
fat appears to be increased even in normal-weight PCOS [65,
66]. Additionally, even when not increased, the abdominal fat of
normal-weight women with PCOS is comprised of a more significant
proportion of small dysfunctional adipocytes (hyperplasia),
compared with what is found in controls [8]. In overweight/
obese women with PCOS, visceral adipocytes are enlarged
(hypertrophic), expanded, and dysfunctional [39, 50, 67], and they positively correlate with T, free testosterone, and A4 levels [8].
Conversely, some studies did not show more significant visceral
fat accumulation in either obese or nonobese PCOS [68, 69];
nevertheless, there is a global excess of fat mass in PCOS [68, 70].
Despite persistent debate regarding the presence of increased
visceral fat in women with PCOS, these women have decreased
gluteal-femoral gynaecoid adiposity.
Subcutaneous adipose tissue in PCOS
Adipocytes from various regions produce different signaling
molecules that influence insulin secretion and glucose and lipid
metabolism in muscle and liver [71]. In vitro, T reduces early-
stage adipocyte differentiation, limiting adipocyte number
and fat storage in abdominal subcutaneous adipose tissue of
lean women with PCOS [72]. When pronounced, abdominal subcutaneous adipose tissue is associated with hyperandrogenemia
and a higher risk of comorbidities [73-75]. In PCOS, subcutaneous
adipocytes may also be enlarged or hyperplasic with limited capacity
to store fat, thus favoring increased release of FFAs with
uptake in lean non-PCOS cells and omentum. This lipotoxicity
explains insulin resistance in lean, non-PCOS individuals [66, 76].
Furthermore, SAT increased conversion of the weak androgen
(A4) into the potent androgen (T), and enhanced local adipogenesis
within adipocytes predisposes to fatty acid overspill into
the systemic circulation in non-PCOS and normal-weight PCOS
[77]. Abnormal subcutaneous adipocytes (hyperplasic or hypertrophic)
decrease insulin-mediated glucose utilization, reduce
glucose transporter type 4 expression (GLUT-4), and stimulate
lipolysis through β2-adrenergic receptors, decreasing the HSL
and protein kinase A (PKA) regulatory components [50].
In obese or nonobese PCOS, adipocytes of SAT are equally
insulin-resistant in regard to glucose metabolism and antilipolytic
activity [78]. In preadipocytes of SAT of nonobese PCOS, T
decreases the number of β-adrenergic receptors and diminishes
HSL activity with a lower catecholamine lipolytic effect [47, 50].
The reduced lipolysis in SAT may explain adipocyte hypertrophy
and dysfunction in this depot. Of note, even in normal concentrations,
T amplifies the ability of catecholamines to activate
hydroxylation of triglycerides in FFAs and glycerol [30].
Visceral adipose tissue in PCOS
Among the relevant comorbidities of women with PCOS, about
70% are overweight or obese [78]. Androgen binding capacity
is more significant in VAT [30]. Hyperandrogenemic states such
as obese PCOS may favor visceral deposition even when compared
with obese non-PCOS [65, 79, 80]. Despite previous reports of
women with PCOS having global adiposity rather than visceral
adiposity, most studies showed that adipose tissue has a preferentially
visceral distribution in this syndrome [81]. The preferential
VAT distribution in nonobese PCOS is accompanied
by an increased number of small subcutaneous adipocytes [8].
The increased visceral adiposity in nonobese and obese PCOS
is associated with metabolic dysfunctions such as dyslipidemia,
abnormal carbohydrate metabolism, insulin resistance, arterial
hypertension, low-grade chronic inflammation, and a
pro-thrombotic state, resulting in a higher risk of T2DM and
CVD [18, 51, 82-84]. Of note, preferential central obesity distribution
in women with PCOS was not found [85].
VAT releases more FFAs than SAT [86], suggesting that there
are regional variations in adipocyte lipolysis. Thus, VAT has
higher catecholamine-stimulated lipolysis, lower lipoprotein
lipase activity, and lower glucose uptake than SAT [71, 87]. Because
VAT is linked to the liver via the portal vein, alterations
in visceral adipocytes directly affect liver function. VAT lipolysis
is enhanced in PCOS, while the lipolysis in abdominal SAT
is diminished by T in this syndrome [47, 51]. The increased rate
of VAT lipolysis in PCOS, even in nonobese women, is due to
β2-adrenoceptor catecholamine-induced adipocyte lipolysis
mediated by a unique stoichiometric change in the properties
of the PKA-HSL enzyme complex [47]. Additionally, in in vitro
studies, T was shown to be a positive contributory factor to catecholamine lipolytic capacity. In summary, human and animal
data strongly suggest unique upregulation in visceral fat cells
due to selective increases in the function of the PKA-HSL complex
[88]. The altered lipolysis in both SAT and VAT in women
with PCOS has an important pathophysiological role.
Hyperandrogenism, obesity, and insulin resistance in PCOS
In women with PCOS, an increase in luteinizing hormone
pulsatility is associated with a decrease in follicle-stimulating
hormone levels, and this combined effect on ovarian theca and
granulosa cells results in increased production and release of T
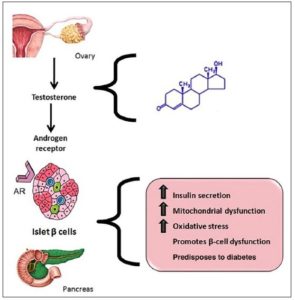
and A4. Higher testosterone levels directly stimulate the secretion
of insulin by pancreatic β-cells [89-93] (Fig. 2). Additionally,
higher circulating insulin levels amplify the effects of luteinizing
hormone (LH) on theca cells and on T production, which is
increased by LH. Furthermore, the increase in T promotes not
only pancreatic β-cell dysfunction, but also the expression and
accumulation of adipose tissue in the omentum and intra-abdominal
organs [94].
Hyperandrogenism, obesity, and insulin resistance in PCOS
In women with PCOS, an increase in luteinizing hormone
pulsatility is associated with a decrease in follicle-stimulating
hormone levels, and this combined effect on ovarian theca and
granulosa cells results in increased production and release of T
and A4. Higher testosterone levels directly stimulate the secretion
of insulin by pancreatic β-cells [89-93] (Fig. 2). Additionally,
higher circulating insulin levels amplify the effects of luteinizing
hormone (LH) on theca cells and on T production, which is
increased by LH. Furthermore, the increase in T promotes not
only pancreatic β-cell dysfunction, but also the expression and
accumulation of adipose tissue in the omentum and intra-abdominal
organs [94].
As previously stated, in visceral adiposity, adipocytes are
enlarged, and insulin resistance and increased lipolysis occur
[95]. More significant release of FFAs in the portal vein affects
liver function [47, 50]. Although hypertrophic adipocytes are markers
of metabolic derangements, dysregulated metabolism in
PCOS can be related to either larger or numerous small adipocytes
of abdominal subcutaneous adipose tissue [66]. In clinical
studies, androgens strongly correlated with markers of central
adiposity [7, 18, 96].
Androgens can induce insulin resistance by directly affecting
skeletal muscle and adipocytes through increased adipocytokine
secretion and increased visceral adiposity in experimental
studies [97-99]. Even normal-weight women with PCOS exhibited
increased fasting insulin levels and greater pancreatic β-cell
response than body mass index-matched controls [8]. Of note,
visceral fat deposition is primarily driven by insulin resistance,
and VAT distribution exacerbates insulin resistance. This bidirectional
vicious cycle is present in PCOS [79]. The hypertrophic
insulin-resistant adipocyte appears to be a consequence of diminished
insulin-dependent autophosphorylation [100], reduced
IRS-2 tyrosine phosphorylation, or reduced GLUT-4 within the
adipocyte [101, 102]. In summary, higher androgen levels can impair
the insulin effect directly or through various changes in different
tissues [103]. To overcome peripheral insulin resistance, more insulin
is produced by β-cells, resulting in hyperinsulinemia. This
hyperinsulinemia, acting on theca cells, increases testosterone
synthesis. Furthermore, chronic androgen excess predisposes to
T2DM secondary to pancreatic β-cell failure [93, 104, 105].
Conclusions
There is a close interrelationship between androgens and adipocytes
in women with PCOS. Adipocytes produce potent androgens, and androgens modulate adipocyte proliferation, hypertrophy, macrophage invasion, and overproduction of various adipocytokines that stimulate steroidogenic cell secretion.
There is a role for hyperandrogenism in adipose tissue distribution,
favoring subcutaneous abdominal wall and visceral adiposity instead of fat distribution to the hips and thighs. In nonobese women with PCOS, adipocytes can be hyperplastic, enlarged, and dysfunctional in subcutaneous adipose tissue, and this finding may explain the insulin resistance and abnormal lipid metabolism with lipid toxicity in this group. The intra-abdominal deposition also appears to be increased in lean PCOS. Overweight/obese women with PCOS have a global excess
fat mass. When pronounced, abnormal subcutaneous adipose
tissue is also associated with hyperandrogenism and a higher
risk of comorbidities. However, in obese PCOS, obesity is preferentially centralized, even when compared with body mass index-matched controls. In PCOS with obesity, the hypertrophic adipocytes are also associated with dyslipidemia, abnormal carbohydrate metabolism, insulin resistance, type 2 diabetes mellitus, low-grade chronic inflammation, the pro-thrombotic state, and cardiovascular disease. Taken together, these findings reinforce the notion that lowering fat mass is an essential therapeutic measure in the management of women with PCOS, particularly when overweight or obese.
Acknowledgment:
The authors are grateful to Philip Lindeman, MD, PhD, Liberty Medical Communications, for the English revision.
Conflict of interest
The authors declare there are either no financial or other conflicts of interest that could be perceived as prejudicing the impartiality of this study.