Introduction
In 1942, Fuller Albright et al. described a disorder characterized by end-organ resistance to parathyroid hormone (PTH) resulting in increased serum PTH levels, hypocalcemia and hyperphosphatemia. The condition was named ‘pseudohypoparathyroidism’ (PHP) given that the PTH was elevated in the face of low calcium and high phosphorus levels [1]. These patients had specific somatic and developmental abnormalities such as round facies with a ‘short, thick figure’, heterotopic subcutaneous ossifications (SCOs), brachydactyly and cognitive impairment [2,3]. This spectrum was later to be termed PHP type 1A (PHP1A). Patients with PHP1A have GNAS mutations on the maternally inherited allele and manifest resistance to multiple Gs protein coupled hormones (e.g. PTH, thyroid-stimulating hormone [TSH], luteinizing hormone [LH], follicle-stimulating hormone [FSH], growth hormone-releasing hormone [GHRH]) due to paternal imprinting of Gαs (Galpha(s)) transcripts in specific tissues [4]. The prevalence of PHP has never been estimated. However, it seems to be extremely rare, as fewer than 60 cases have been reported until 2016 worldwide [5,6]. We present a female patient with PHP, who addressed to us in her late twenties due to extreme obesity and menstrual irregularities.
Case report
The patient is a 28-year-old female referred to the Clinic for Endocrinology due to irregular cycles, obesity, and hair loss. Previously in 2014 she had an exploratory curettage due to prolonged bleeding that resulted in endometrial hyperplasia upon histopathological examination. Her menarche was at age 13, after which her menstruations have always been irregular according to the type of oligomenorrhea, with the longest period without menstruation being of 3 months. In childhood, she was examined for obesity under the suspicion of hypercortisolism, which was ruled out. She gained weight in early childhood, with a maximum of 120 kg at the age of 12. Psychological testing in childhood concluded low intellectual capacity (mild mental retardation). She took no medicines at that time. Her mother had diabetes mellitus and had three pregnancies, the first twin pregnancy ended in a spontaneous abortion at 6 months, while the second child died at 3.5 years old of convulsions.
Clinical findings of the present case were: weight 97 kg, height 148 cm, body mass index (BMI) 44.2 kg/m², waist circumference 125 cm, hip circumference 138 cm. She was obese with phenotypic characteristics of AHO (Albright hereditary osteodystrophy): brachydactyly, short stature, stocky habitus, macrocephaly, and round face. There were no signs of hirsutism. Examination of the head, neck, and chest were in order. Blood pressure was 140/90 mmHg and heart rate 80 beats per min. She displayed short fingers on both hands, except for thumbs and had palpable subcutaneous ossifications in the pretibial region of the lower extremities.
Laboratory tests were: Ca 2.35 mmol/L, ionized Ca 1.21 mmol/L, PO4 1.16 mmol/l, Na 140 mmol/L, K 4.7 mmol/L, PTH 217 ng/L, Vitamin D 8.1 ng/mL, ACTH 6.1 ng/L, Cortisol 08h 187 nmol/L, Cortisol 20h 124 nmol/L, Cortisol 24h 62.9 nmol/L, TSH 6.3 mIU/L, FT4 9.0 ng/L, TG-AB and TPO-AB were negative, calcitonin 40.1 ng/L, FSH 12.7 IU/L, LH 4.6 IU/L, E2 176.8 pmol/L, Progesterone 2.7 nmol/L, prolactin 319.6 mIU /L, testosterone 0.92 nmol/L, 17OHPg 1.4 nmol/L, DHEAS 1.0 µmol/L, SHBG 31.8 nmol/l, AMH 0.543 ng/mL, IGF-1 179.4 ng/mL, GH 0.88 mIU/L, Karyotype 46, XX, 24-hour urine calcium and phosphorus levels were in normal range. OGTT glycemia: 4.4; 6.9; 5.5; 5.8 and 5.7 mmol/L. Insulin: 17.1; 67.4; 48.8; 50.1 and 40.1 mIU/L. HOMA index was 3.3.
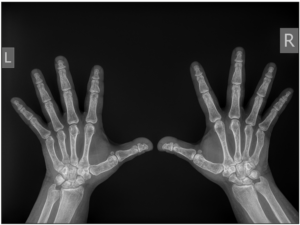
Chest radiography, neck ultrasound, and abdominal ultrasound were normal. Breast ultrasound showed cystic fibrodysplasia. Hand radiography showed shorter metacarpal bones of the fourth finger bilaterally with discrete degenerative changes (Figure 1). Cranial radiography found no osteolytic or osteoblastic changes. Ophthalmological examination found no signs of lenticular opacity. Dual X ray absorptiometry showed normal bone density, Z score L1-L4= +2.2; left hip total Z score= 0.0; and whole-body scan: Z score= +1.2. Total body fat on whole body scan was 44.5%.
Neuropsychological assessment revealed mild deficit in cognition, adaptive behavior and executive function. The patient was prescribed with thyroxin, calcitriol, metformin, diet regimen and regular visits were scheduled in 6 months.
Discussion
In 1942, Fuller Albright et al. described a disorder characterized by end-organ resistance to PTH resulting in increased serum PTH levels, hypocalcemia and hyperphosphatemia. These patients lacked the appropriate response to the administration of PTH and had blunted urinary cAMP and phosphate excretion. The condition was named PHP given that the PTH was elevated in the face of low calcium and high phosphorous levels [1]. These patients had specific somatic and developmental abnormalities such as a round facies with a ‘short, thick figure’, heterotopic SCOs, brachydactyly and cognitive impairment [1-3]. Later this entity was named PHP1A. Approximately a decade later, Albright et al. also identified a patient who had many of these same physical features but normal calcium, phosphorous and PTH levels as well as a normal phosphaturic response to PTH; this was named pseudopseudohypoparathyroidism (PPHP). The physical phenotype for both PHP1A and PPHP was termed Albright hereditary osteodystrophy (AHO). AHO is a disorder caused by heterozygous inactivating mutations affecting exons 1–13 of GNAS, the gene encoding the α-chain of the stimulatory G protein (Galpha[s]: Gαs), which links receptors for many hormones and neurotransmitters to activate adenylyl cyclase [7]. The locus is controlled by genomic imprinting such that transcription from one parental allele is suppressed, often only partially. This imprinting is regulated through differentially methylated regions that are in the promoter regions for each exon (except exon 1 of GNAS needed for Gαs) [2,3,8,9]. Patients with PHP1A have GNAS mutations on the maternally inherited allele and manifest resistance to multiple Gs protein coupled hormones (e.g. PTH, TSH, LH, FSH, GHRH) due to paternal imprinting of Gαs transcripts in specific tissues. These patients often have severe obesity, especially of early-onset. Patients with PPHP have GNAS mutations on the paternally inherited allele and have the AHO phenotype alone without hormonal resistance or the severe obesity. The identification of the difference in the obesity phenotype between PHP1A and PPHP raised the possibility that paternal imprinting in the hypothalamus may be the cause [4]. Thus, women affected with AHO have children with PHP1A affected by hormonal resistance and obesity, whereas men with AHO have children with PPHP without hormonal resistance and obesity [2-4,10]. Although the phenotype of a patient with AHO can be explained by the parental mode of transmission of the GNAS mutant allele, spontaneous mutations can also occur. In addition, abnormalities in imprinting due to mutations that affect methylation patterns typically cause a condition termed PHP1B. PHP1C is also within this group of disorders but difficult to prove, as it presents in the same manner as PHP1A [11].
A molecular cause can be identified in an estimated 80–90% of patients with PHP or related disorders [12,13]. The most common underlying mechanisms are de novo or autosomal dominantly inherited genetic mutations and/or epigenetic, sporadic or genetic-based alterations, within or upstream of GNAS, PRKAR1A, PDE4D or PDE3A [14-19]. In the absence of molecular analysis, the clinical and biochemical overlap between PHP and related disorders can lead to challenges in diagnostic classification and thus in understanding of the natural history of the different types. A consensus meeting was organized to develop recommendations for the diagnosis and management of patients with PHP and related disorders [20].
Our patient revealed main clinical features of PHP1A, as decreased growth velocity, early onset obesity, brachydactyly, stocky habitus, round face, PTH, TSH, gonadotropin resistance, and neurocognitive impairment. The participants in the First International Consensus Statement on PHP agreed that the diagnosis should be based on major criteria, including resistance to PTH, brachydactyly, early onset obesity, ectopic ossifications, and cognitive impairment [20].
PHP and related disorders vary considerably in clinical presentation and disease severity between affected individuals, even among patients carrying the same genetic alteration. The clinical symptoms (e.g. ossifications and brachydactyly) and abnormalities that can be detected in a laboratory (e.g. hypocalcemia and raised PTH levels) typically worsen during mid and late childhood and are usually unnoticed in very young children. A correct diagnosis can thus be elusive during infancy and in patients with atypical features [21].
The term AHO is used to indicate a constellation of physical features originally described by Albright, including a round face, a stocky habitus with short stature, brachydactyly and ectopic ossification [1]. Short bones are not present at birth and result from premature closure of the epiphyses, leading to a reduced period of growth. Although all bones tend to be short, shortening is most marked acrally (hands and feet). Subsequently, developmental delay was added as an additional feature of AHO [22,23]. Obesity, particularly of early onset, and macrocephaly relative to height, might be part of AHO which was a feature in presented patient [9,24,25].
In patients with PHP1A, resistance to PTH is usually absent at birth and evolves over life (from 0.2 years to 22 years) while the clinical manifestations typically occur later [14,26,27]. These data suggest that PTH resistance begins in early childhood, and the resultant changes in serum levels of calcium and phosphorus develop gradually, at some point during adulthood [26,28-31]. The first biochemical abnormalities to become apparent are elevated serum levels of PTH and elevated serum levels of phosphorus, followed by hypocalcemia. When hypocalcemia is present, urine levels of calcium are low, whereas calcitriol levels might be either low or normal [14]. An interval of up to 4.5 years usually occurs between the start of the elevation in levels of PTH and phosphorus and the onset of hypocalcemia [30].
Disorders caused by molecular alterations of the GNAS gene or locus can feature ectopic ossification. Ectopic ossification is not calcification and is unrelated to serum levels of calcium and phosphorus. The ectopic ossifications are a manifestation of Gαs deficiency in mesenchymal stem cells, with de novo formation of extraskeletal osteoblasts that form islands of ectopic bone in the dermis and the subcutaneous fat as a result of the differentiation of adipose - derived progenitor mesenchymal stem cells that lead predominantly to web - like intramembranous ossifications [32,33].
In PHP and related disorders, brachydactyly can be classified as type E, which is defined as variable shortening of the metacarpals with, usually, normal length of phalanges, occasionally accompanied by relatively shortened metatarsals. In PHP and related disorders, the fifth, fourth and third metacarpal and the first and fourth distal phalanges are the most affected bones of the hand [5]; metatarsals are often shortened as well. Brachydactyly develops over time and might not be evident in early life [16,34,35]. However, brachydactyly is not specific to PHP and related disorders and can be found in patients with, for example, tricho–rhino–phalangeal syndrome, brachydactyly mental retardation syndrome or Turner syndrome [20].
Patients with PHP1A frequently present with raised serum levels of TSH and thyroid hormone levels that are normal or slightly reduced. Some patients present with overt clinical hypothyroidism. The literature does not contain specific data on how to treat TSH resistance in patients with PHP and related disorders. Statement should be the same as those used in any other form of hypothyroidism or subclinical hypothyroidism. In general, patients should be screened for autoimmune thyroid disease. Nevertheless, because autoimmune thyroid disease is highly prevalent, the presence of auto immunity will not rule out concomitant TSH resistance in a given patient [36].
Resistance to gonadotropins seems to be less severe than resistance to other hormones, such as PTH and TSH; laboratory abnormalities such as elevated levels of LH or FSH have been reported by some groups, whereas others have not been able to confirm these findings [37-46]. These findings have led to the hypothesis that patients with PHP1A display only partial resistance to gonadotropins [41]. The pubertal growth spurt might be blunted or absent in both girls and boys with PHP1A. Although the basis for this defect is unknown, it could be related to insufficient sex steroid production or premature epiphyseal closure [41]. Systematic data are scarcer for PHP1B and PPHP than for PHP1A, but these patients are thought to have normal gonadal function [47]. Similar to PHP1A, variable resistance to gonadotropins has been described in patients with acrodysostosis and mutations in the PRKAR1A gene, as well as in anecdotal reports of single patients with mutations in PDE4D [48,49]. Furthermore, despite basal gonadotropin and sex steroid levels that are within normal limits, menstrual irregularities seem to be common among female patients with PHP1A [41]. Unassisted and uneventful pregnancies have been reported in female patients with PHP1A [41,50]. These pregnancies are more often seen in women with PPHP, who give birth to offspring with PHP1A [22,38,42,50-52]. In a few cases, either infertility or the need for assisted reproductive technology to obtain pregnancy have been reported [53-55]. No data are available on lactation, but there are no specific reports that these patients are unable to lactate.
Long-term treatment of hypocalcemia associated with PTH resistance is similar but usually more insistent than that of primary hypoparathyroidism, with the use of active vitamin D metabolites (calcitriol) or analogues (alphacalcidol) and oral calcium supplements as and when required [56]. The current approach is to reduce serum levels of PTH to the upper portion of the reference range to avoid the suppression of PTH, which can be associated with hypercalciuria and renal calcification. Serum levels of PTH that are at the upper limits of the reference range are sufficient to enhance calcium reabsorption in the distal renal tubule, thus helping to prevent hypercalciuria [27]. However, PTH levels should not be too high, as long - standing PTH excess might have adverse effects on skeletal mineralization or on the growth plate [57-60].
Chronic hypocalcemia and associated hyperphosphatemia can result in intracranial deposition of calcium, a feature usually referred to as Fahr syndrome [61]. These calcifications occur predominantly in the basal ganglia but might extend widely to the thalamus and the cortex. This form of ectopic calcification is due to elevated levels of the calcium–phosphorus product and hence, has not been described in patients with PPHP or progressive osseous heteroplasia (POH) or those with a mutation in the PRKAR1A or PDE4D genes [16,17,62-65]. Other ectopic depositions of calcium and phosphorus occur in the eyes, which lead to cataracts (peripheral lenticular opacities). Corneal opacities, macular degeneration, nystagmus, anisocoria, tortuosity of retinal vessels and microphthalmia have also been reported [62,66-68].
The prevalence of osteoporosis in patients with PHP and related disorders is unknown. In these patients, bone loss might occur as a result of untreated hypogonadism, long- term excess levels of PTH, GH deficiency and/or the onset of physiological menopause. The physiological action of PTH on bone is mainly to promote bone resorption, but the extent to which PTH signaling in bone is defective in patients with PHP and related disorders is not completely clear. The bone remodeling response to PTH, which is independent of vitamin D action, seems to be intact in these PHP patients, which suggests a possible increased risk of osteoporosis in patients with sustained increased levels of PTH despite treatment with calcium and vitamin D. Nevertheless, bone density seems to be normal to high in patients with PHP1A, due to decreased bone turn over, in particular in those who do not have excessive circulating levels of PTH [58,69].
Large scale studies aimed at determining malignancy risk in patients with PHP and related disorders have not been carried out. A Danish study investigated all patients with a diagnosis of PHP or a related disorder (clinical and/or genetically confirmed) to determine their mortality data and risk of complications using data from the Danish National Patient Registry [62]. With a total of 60 cases, patients with PHP or a related disorder were found to have an increased risk of neuropsychiatric disorders, infections, seizures and cataracts, whereas their risk of renal, cardiovascular and malignant disorders and fractures was similar to that of the general background population [20].
The prognosis of PHP is variable. In mild forms of the disease, when treated appropriately with calcium and vitamin D, a normal life expectancy is not unreasonable. For others with a more severe expression of the AHO phenotype, the presence of obesity, sleep apnea, and mental retardation can cause significant morbidity and mortality [62].
Conclusion
Patients with PHP and related disorders face a wide range of problems from early childhood to adulthood. These include potentially severe alterations in mineral metabolism, which could be associated with seizures, other endocrine deficiencies due to hormone resistance that lead to hypothyroidism, hypogonadism and GH deficiency, growth impairment independently of hormonal status, ectopic ossifications, skeletal issues and cognitive and psychomotor impairment. This highly heterogeneous clinical picture requires a mandatory multidisciplinary approach and specialized expertise to manage each of clinical aspects and potential complications of PHP through every stage of life.